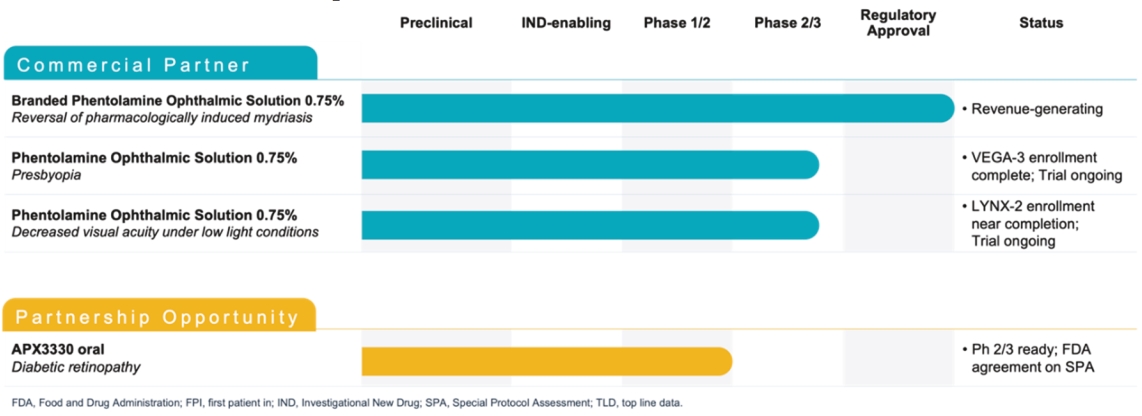
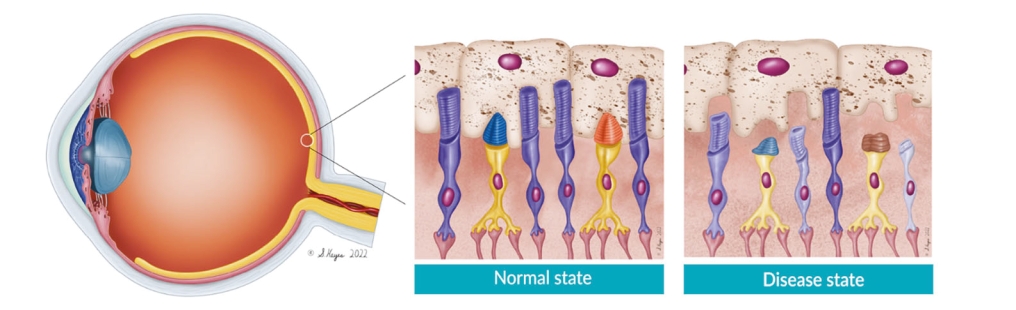
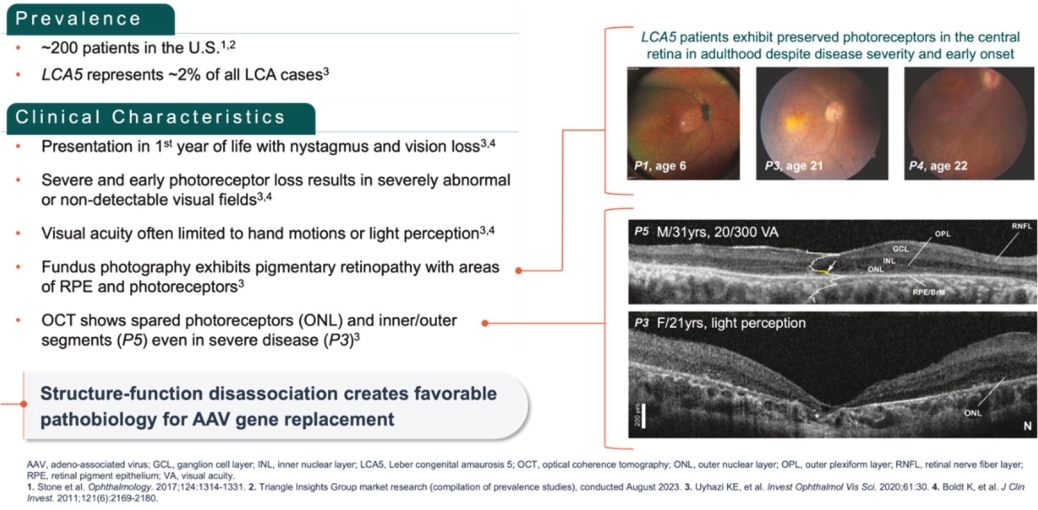
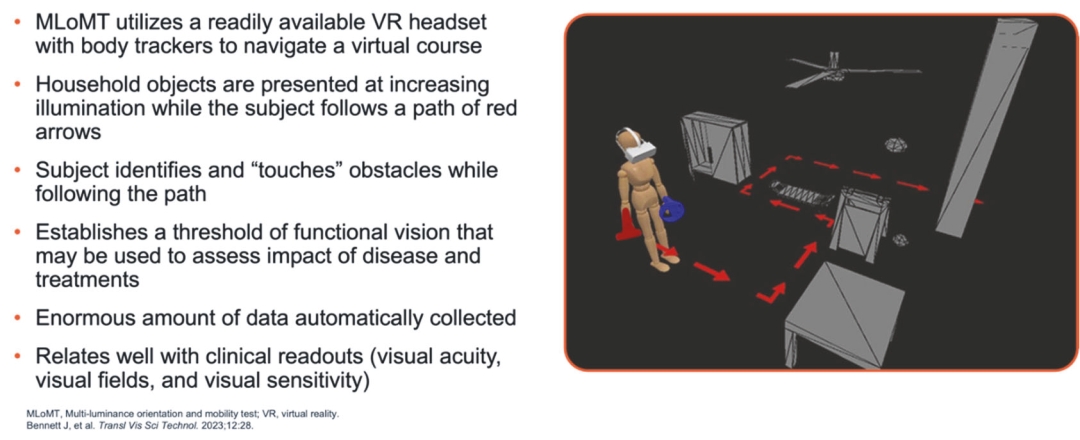
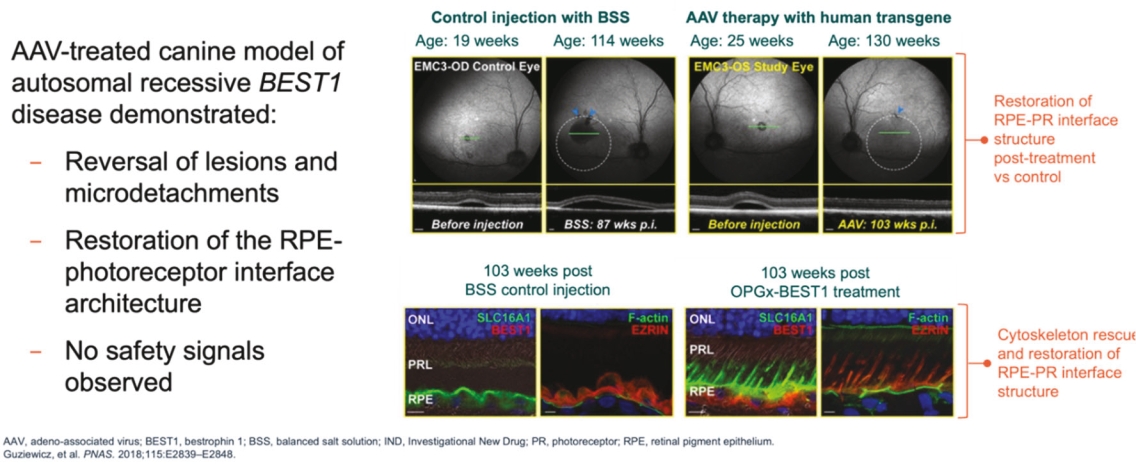
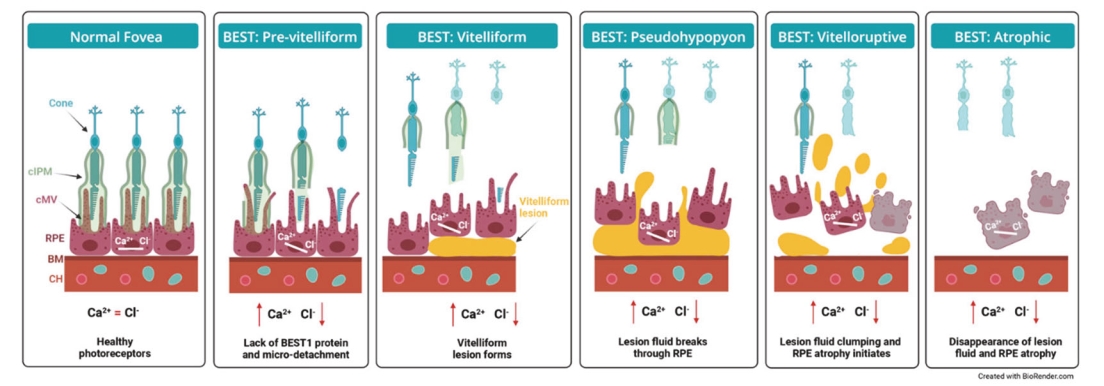
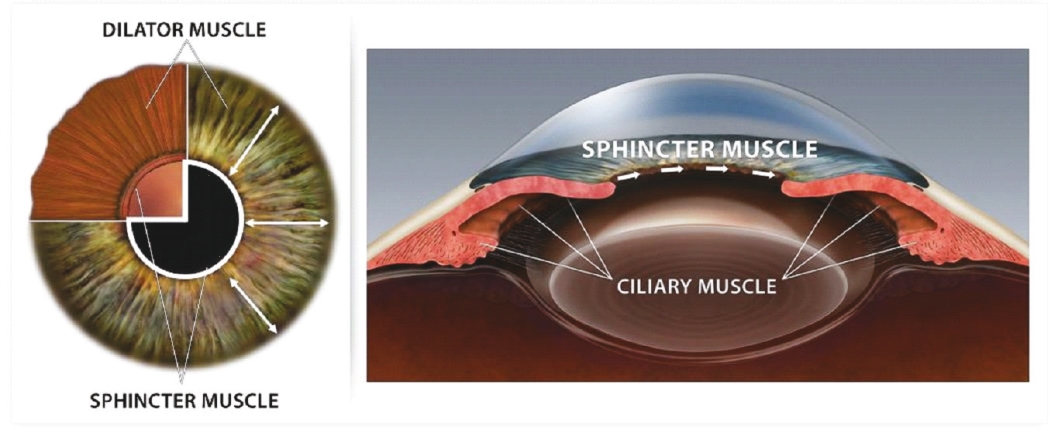
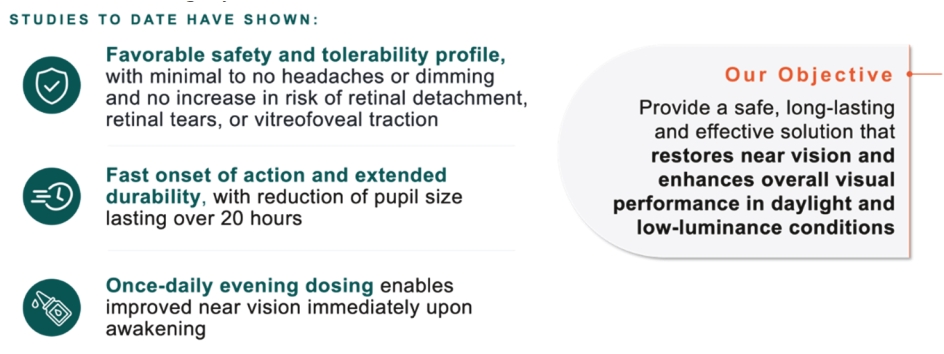
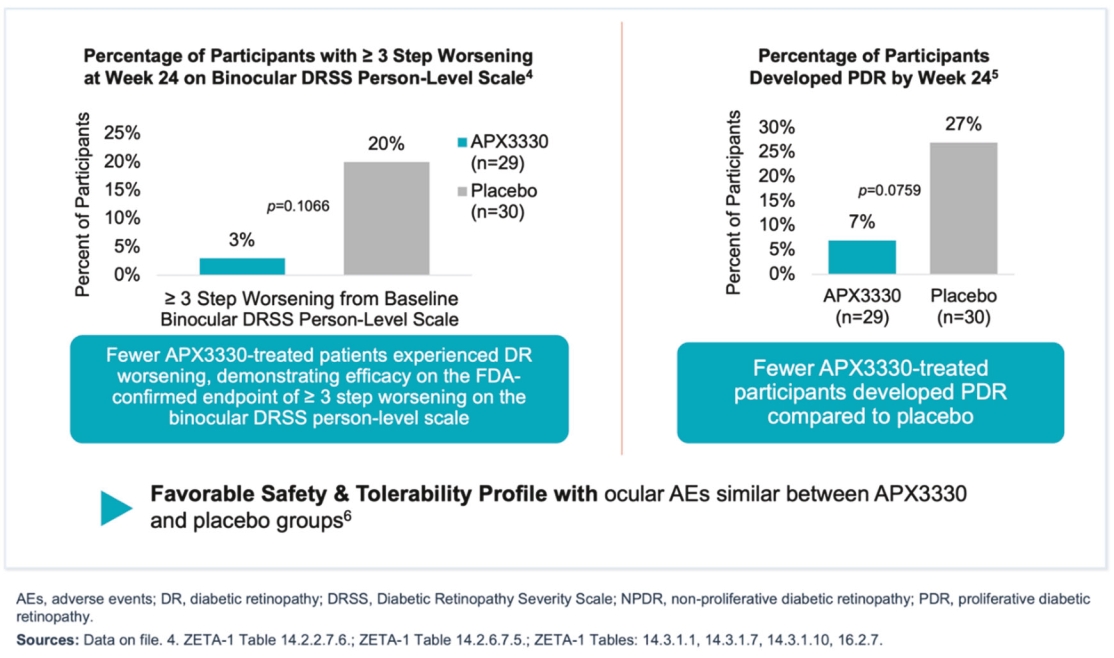
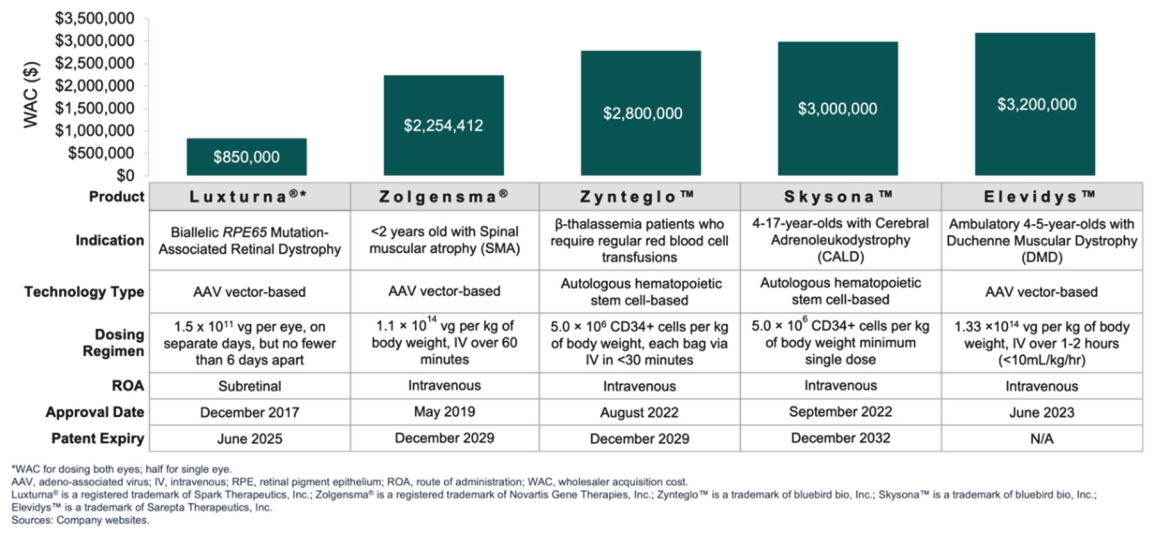
Filed Pursuant to Rule 424(b)(5)
Registration No. 333-276462
The information contained in this preliminary prospectus supplement is not complete and may be changed. We may not sell these securities or accept an offer to buy these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus supplement is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where such offer or sale is not permitted.
Subject to Completion, dated March 20, 2025
PRELIMINARY PROSPECTUS SUPPLEMENT
(to Prospectus dated January 23, 2024)
Shares of Common Stock
Warrants to Purchase up to Shares of Common Stock
Pre-Funded Warrants to Purchase Shares of Common Stock
Up to Shares of Common Stock underlying the Warrants and Pre-
Funded Warrants
We are offering shares of our common stock and warrants to purchase up to shares of our common stock (the “Warrants”) pursuant to this prospectus supplement and the accompanying prospectus. Each share of our common stock is being sold together with a Warrant to purchase one share of our common stock. The shares of our common stock and Warrants are immediately separable and will be issued separately, but will be purchased together in this offering. The public offering price for each share of our common stock and related Warrant is $ . Each Warrant will be exercisable immediately at an exercise price of $ per share and will be exercisable immediately upon issuance and will expire on the five year anniversary of the date of issuance. The holder of the Warrant may, at their sole discretion, exercise each of their Warrants for one Pre-Funded Warrant at an exercise price of $ (which is the per share exercise price minus $0.0001)
We are also offering Pre-Funded Warrants (as defined below) to certain purchasers, whose purchase of common stock and Warrants in this offering would otherwise result in the purchaser, together with its affiliates and related parties, beneficially owning more than 4.99% of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if they so choose pre-funded warrants (“Pre-Funded Warrants”) in lieu of the common stock and Warrants that would otherwise result in ownership in excess of 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock. The public offering price for each Pre-Funded Warrant will equal the price per share minus $0.0001, and the exercise price of each Pre-Funded Warrant will be $0.0001 per share of our common stock. The Pre-Funded Warrants offered hereby will be immediately exercisable and may be exercised at any time until exercised in full.
For each Pre-Funded Warrant we sell, the number of shares of common stock we are offering will be decreased on a one-for-one basis. Because we will issue a Warrant with the issuance of each share of common stock or Pre-Funded Warrant, the number of Warrants sold in this offering will not change as a result of a change in the mix of the shares of common stock and Pre-Funded Warrants sold.
We are also offering the shares of our common stock that are issuable from time to time upon exercise of the Warrants and the Pre-Funded Warrants.
We refer to the shares of our common stock, the Warrants, the Pre-Funded Warrants and the shares of our common stock issued or issuable upon exercise of the Warrants and Pre-Funded Warrants, collectively, as the “Securities.”
Our common stock is listed on the Nasdaq Capital Market (“Nasdaq”) under the symbol “IRD”. The last reported sale price of our common stock on Nasdaq on March 19, 2025 was $1.15 per share. There is no established trading market for the Warrants or the Pre-Funded Warrants, and we do not expect a market to develop. We do not intend to apply to list the Warrants or the Pre-Funded Warrants on any securities exchange or other nationally recognized trading system. Without an active trading market, the liquidity of the Warrants and the Pre-Funded Warrants will be limited.
We are a “smaller reporting company” as defined under federal securities laws and as such, have elected to comply with reduced public company reporting requirements. See “Prospectus Supplement Summary — Implications of Being a Smaller Reporting Company.”
Investing in our common stock involves significant risks that are described in the “Risk Factors” section beginning on page S-52 of this prospectus supplement and page 7 of the accompanying prospectus, and in the other documents that are incorporated by reference herein. You should read the entire prospectus supplement and the accompanying prospectus, including any information incorporated by reference herein or therein, carefully before you make your investment decision.
|
Per Share
and Related
Warrant
|
Per Share
and Related
Pre-Funded
Warrant
|
Total |
|||||||
Public offering price
|
$ |
$ |
$ |
||||||
Underwriting discounts and commissions(1)
|
$ |
$ |
$ |
||||||
Proceeds, before expenses, to us(2)
|
$ |
$ |
$ |
(1) |
Includes an underwriting discount of 6.0% on the public offering price. See the section titled “Underwriting” beginning on page S-106 of this prospectus supplement for a description of the compensation payable to the underwriter. |
(2) |
The amount of offering proceeds to us presented in this table does not give effect to the exercise, if any, of the Warrants or the Pre-Funded Warrants being issued in connection with this offering. |
Neither the Securities and Exchange Commission (“SEC”) nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus supplement or the accompanying base prospectus. Any representation to the contrary is a criminal offense.
The underwriter expects to deliver the shares of common stock, Warrants and Pre-Funded Warrants against payment therefor on or about , 2025.
Sole Managing Underwriter
Craig-Hallum
Prospectus supplement dated , 2025