EXHIBIT 99.1
Published on
Exhibit 99.1

Opus Genetics R&D Science Forum June 16, 2026 | 10:00 AM – 12:00 PM ET

Certain statements contained in this presentation are not statements of
historical fact and are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements give current
expectations or forecasts of future events or our future financial or operating performance. Such statements include, but are not limited to, statements concerning our data from and future enrollment for our clinical trials, our pipeline of
additional indications and anticipated regulatory milestones. In some cases, you can identify forward-looking statements by the following words: “aim,” “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,”
“ongoing,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” or the negative of those terms, and similar expressions that convey uncertainty of future events or outcomes to identify these forward-looking statements. These
forward-looking statements reflect our management’s beliefs and views with respect to future events, are based on estimates and assumptions as of the date of this presentation and are subject to risks and uncertainties, many of which are
beyond our control, that could cause our actual results to differ materially from those in these forward-looking statements, including, without limitation: our gene therapy product candidates are based on a novel technology and manufactured
by a third party, which may result in delays and difficulties in obtaining regulatory approval; our planned clinical trials may face substantial delays, result in failure, or provide inconclusive or adverse results that may not satisfy U.S.
Food and Drug Administration (“FDA”) requirements to further develop our therapeutic products; delays or difficulties associated with patient enrollment in clinical trials may affect our ability to conduct and complete those clinical trials
and obtain necessary regulatory approvals; changes in regulatory requirements could result in increased costs or delays in development timelines; we depend heavily on the success of our product pipeline; if we fail to find strategic partners
or fail to adequately develop or commercialize our pipeline products, our business will be materially harmed; others may discover, develop, or commercialize products similar to those in our pipeline before or more successfully than we do or
develop generic variants of our products even while our product patents remain active, thereby reducing our market share and potential revenue from product sales; we do not currently have any sales or marketing infrastructure in place and we
have limited drug research and discovery capabilities; the future commercial success of our products could significantly depend upon several uncertain factors, including third-party reimbursement practices and the existence of competitors
with similar products; product liability lawsuits against us or our suppliers or manufacturers could cause us to incur substantial liabilities and could limit commercialization of any product candidate that we may develop; failure to comply
with health and safety laws and regulations could lead to material fines; we have not generated significant revenue from sales of any products and expect to incur losses for the foreseeable future; our future viability is difficult to assess
due to our short operating history and our future need for substantial additional capital, access to which could be limited by any adverse developments that affect the financial markets; raising additional capital may cause our stockholders
to be diluted, among other adverse effects; instability and operational disruptions at government agencies, such as the FDA, may adversely impact our development and commercialization plans by causing delays and requiring the use of
additional, unforeseen resources to obtain regulatory approval for trials or products in our pipeline; we operate in a highly regulated industry and face many challenges adapting to sudden changes in legislative reform or the regulatory
environment, including due to government shutdowns and disruptions at government agencies, which cause delays, requires the use of additional, unforeseen resources, affects our pipeline stability, and could impair our ability to compete in
international markets; we may not receive regulatory approval to market our developed product candidates within or outside of the U.S.; with respect to any of our product candidates that receive marketing approval, we may be subject to
substantial penalties if we fail to comply with applicable regulatory requirements; our potential relationships with healthcare providers and third-party payors will be subject to certain healthcare laws and regulations, which could expose us
to extensive potential liabilities; we rely on third parties for material aspects of our business, such as conducting our nonclinical and clinical trials and supplying and manufacturing bulk drug substances, which exposes us to certain risks;
we may be unsuccessful in entering into or maintaining licensing arrangements or establishing strategic alliances on favorable terms, which could harm our business; inadequate patent protection for our product candidates may result in our
competitors developing similar or identical products or technology, which would adversely affect our ability to successfully commercialize; we may be unable to obtain full protection for our intellectual property rights under U.S. or foreign
laws; we may become involved in lawsuits for a variety of reasons associated with our intellectual property rights, including alleged infringement suits initiated by third parties; we are dependent on our key personnel, and if we are not
successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy; as we grow, we may not be able to operate internationally or adequately develop and expand our sales,
marketing, distribution, and other corporate functions, which could disrupt our operations; the market price of our common stock is expected to be volatile and if we fail to comply with the continued listing standards of Nasdaq, our common
stock may be delisted; and factors out of our control related to our securities, such as securities litigation or actions of activist stockholders, could adversely affect our business and stock price and cause us to incur significant
expenses. We discuss many of these risks in greater detail under Part I, Item 1A “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2025 and in subsequent reports filed with or furnished to the Securities and
Exchange Commission. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on
our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given these uncertainties, you should not place undue
reliance on these forward-looking statements. Any forward-looking statement in this presentation speaks only as of the date hereof or as of the date specified herein. We undertake no obligation to publicly update any forward-looking
statement, whether as a result of new information, future developments or otherwise, except as may be required by applicable laws or regulations. Disclosures and Forward-Looking Statements 2

Meet Our Speakers George Magrath, MD Chief Executive Officer Ben Yerxa,
PhD President Sally Tucker, PhD Chief Medical Officer Ash Jayagopal, PhD, MBA Chief Scientific & Development Officer Joe Schachle, MBA Chief Operating Officer Jean Bennett, MD, PhD University of Pennsylvania Kenneth Fan, MD,
MBA Retina Consultants of Texas Professor Robert MacLaren University of Oxford Lejla Vajzovic, MD Duke University Todd Durham, PhD, MS Foundation Fighting Blindness Opus Genetics Leadership Key Opinion Leaders Bart Leroy, MD,
PhD Ghent University Hospital 3
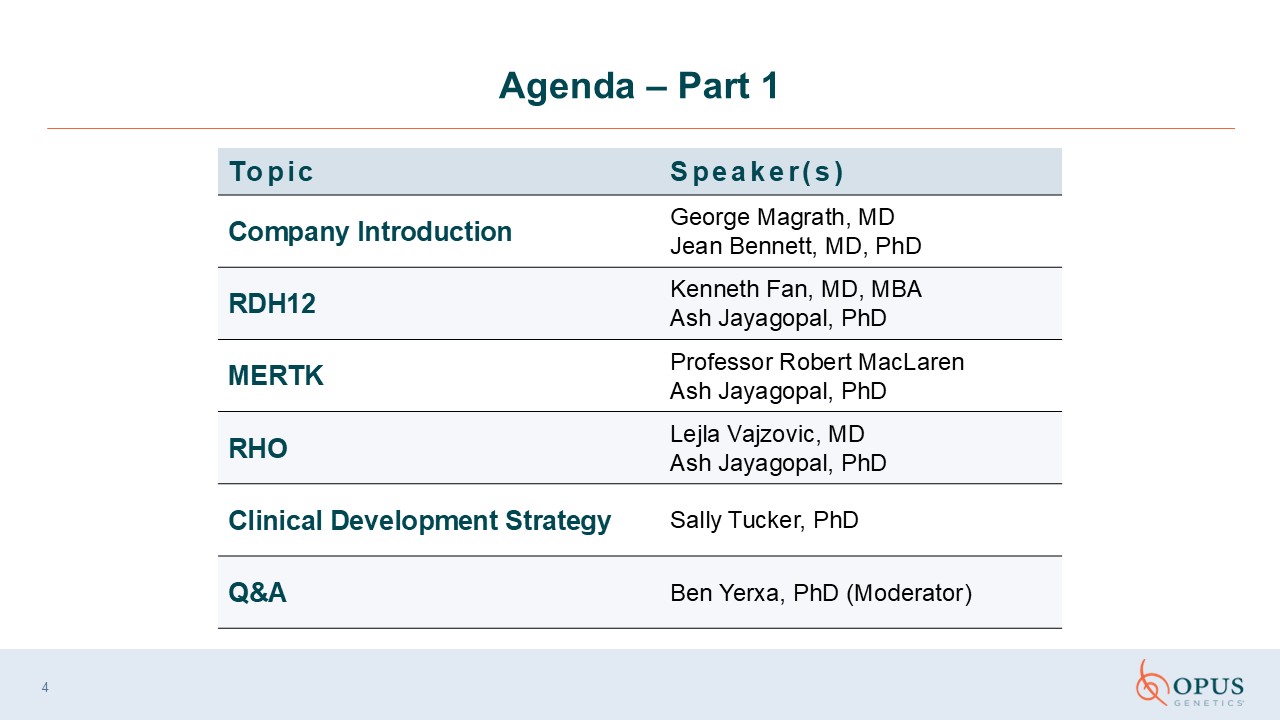
Agenda – Part 1 Topic Speaker(s) Company Introduction George Magrath,
MD Jean Bennett, MD, PhD RDH12 Kenneth Fan, MD, MBA Ash Jayagopal, PhD MERTK Professor Robert MacLaren Ash Jayagopal, PhD RHO Lejla Vajzovic, MD Ash Jayagopal, PhD Clinical Development Strategy Sally Tucker, PhD Q&A Ben
Yerxa, PhD (Moderator) 4

Agenda – Part 2 Topic Speaker(s) LCA5 Bart Leroy, MD, PhD BEST1 Ash
Jayagopal, PhD George Magrath, MD IRD Patient Journey and Disease Prevalence Joe Schachle, MBA Patient Recruitment & Retention Panel Ben Yerxa, PhD (Moderator) Jean Bennett, MD, PhDTodd Durham, PhD, MS Bart Leroy, MD,
PhD Q&A Ben Yerxa, PhD (Moderator) Summary Takeaways George Magrath, MD 5

Company Introduction George Magrath, MD Chief Executive Officer Opus
Genetics
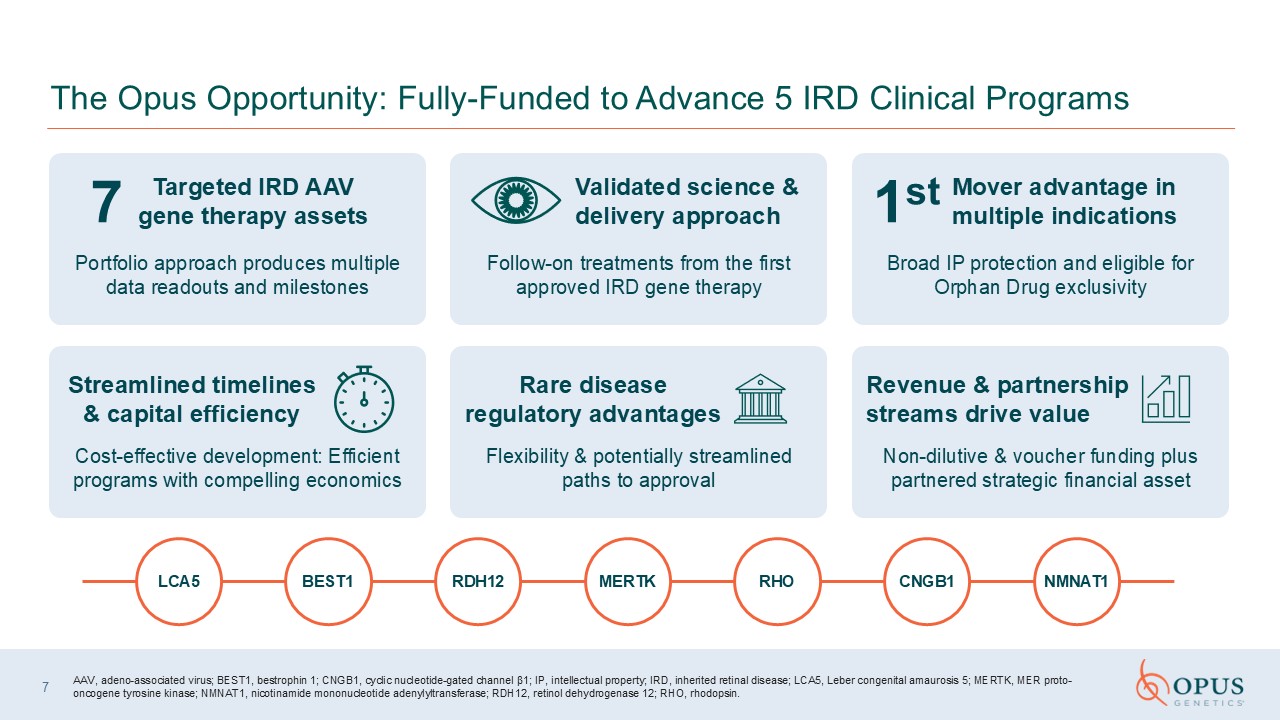
The Opus Opportunity: Fully-Funded to Advance 5 IRD Clinical Programs Portfolio
approach produces multiple data readouts and milestones Follow-on treatments from the first approved IRD gene therapy Broad IP protection and eligible for Orphan Drug exclusivity Cost-effective development: Efficient programs with
compelling economics Flexibility & potentially streamlined paths to approval Non-dilutive & voucher funding plus partnered strategic financial asset Validated science & delivery approach 7 Targeted IRD AAV gene therapy
assets Mover advantage in multiple indications 1st Streamlined timelines & capital efficiency Rare disease regulatory advantages Revenue & partnership streams drive value 7 AAV, adeno-associated virus; BEST1, bestrophin 1;
CNGB1, cyclic nucleotide-gated channel β1; IP, intellectual property; IRD, inherited retinal disease; LCA5, Leber congenital amaurosis 5; MERTK, MER proto-oncogene tyrosine kinase; NMNAT1, nicotinamide mononucleotide adenylyltransferase;
RDH12, retinol dehydrogenase 12; RHO, rhodopsin. LCA5 BEST1 RDH12 MERTK RHO CNGB1 NMNAT1
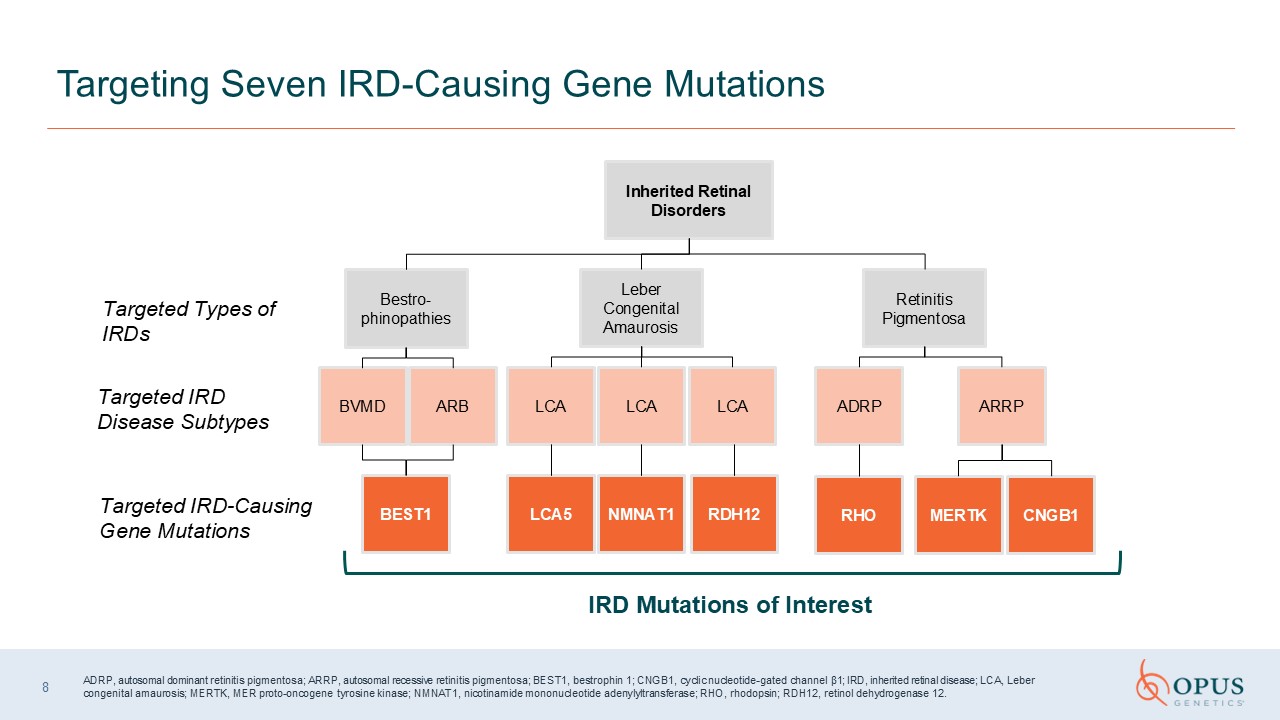
IRD Mutations of Interest Targeting Seven IRD-Causing Gene Mutations Inherited
Retinal Disorders Retinitis Pigmentosa Leber Congenital Amaurosis Bestro-phinopathies ADRP RHO LCA5 NMNAT1 BEST1 BVMD ARB LCA LCA Targeted Types of IRDs Targeted IRD Disease Subtypes Targeted IRD-Causing Gene
Mutations ARRP MERTK CNGB1 LCA RDH12 ADRP, autosomal dominant retinitis pigmentosa; ARRP, autosomal recessive retinitis pigmentosa; BEST1, bestrophin 1; CNGB1, cyclic nucleotide-gated channel β1; IRD, inherited retinal disease; LCA,
Leber congenital amaurosis; MERTK, MER proto-oncogene tyrosine kinase; NMNAT1, nicotinamide mononucleotide adenylyltransferase; RHO, rhodopsin; RDH12, retinol dehydrogenase 12. 8
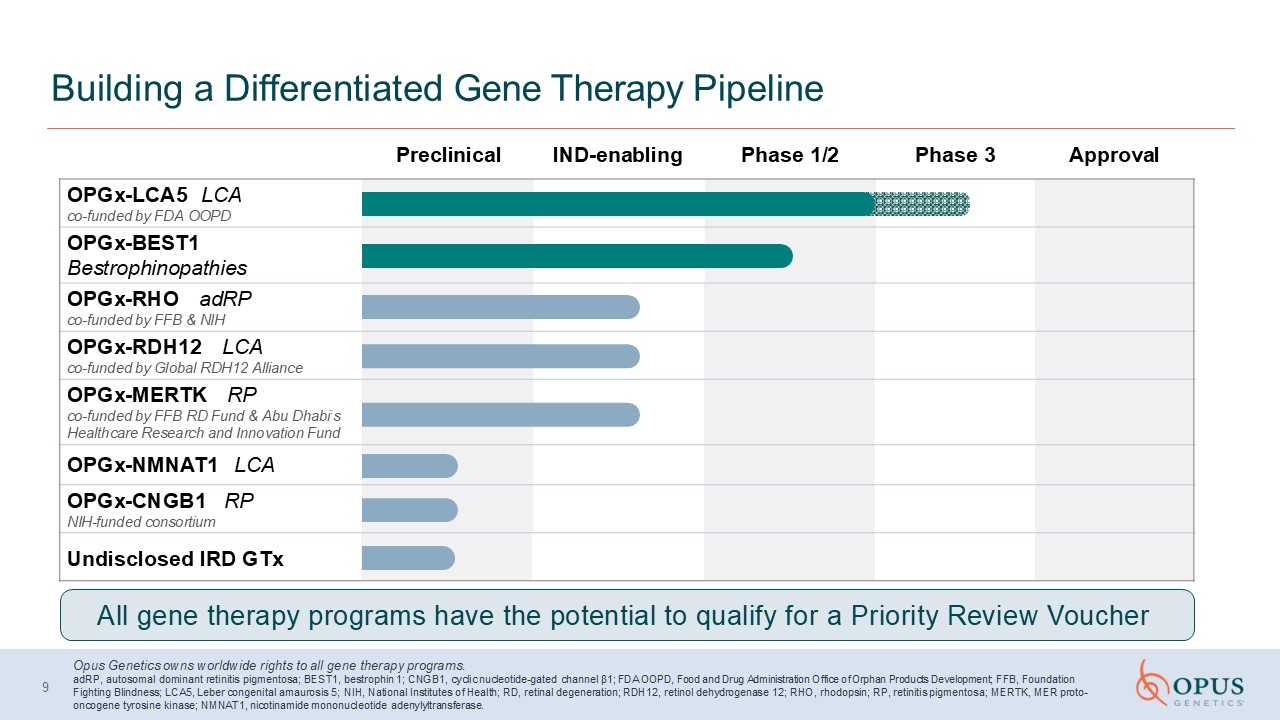
Preclinical IND-enabling Phase 1/2 Phase 3 Approval Building a
Differentiated Gene Therapy Pipeline Opus Genetics owns worldwide rights to all gene therapy programs.adRP, autosomal dominant retinitis pigmentosa; BEST1, bestrophin 1; CNGB1, cyclic nucleotide-gated channel β1; FDA OOPD, Food and Drug
Administration Office of Orphan Products Development; FFB, Foundation Fighting Blindness; LCA5, Leber congenital amaurosis 5; NIH, National Institutes of Health; RD, retinal degeneration; RDH12, retinol dehydrogenase 12; RHO, rhodopsin; RP,
retinitis pigmentosa; MERTK, MER proto-oncogene tyrosine kinase; NMNAT1, nicotinamide mononucleotide adenylyltransferase. OPGx-LCA5 LCA co-funded by FDA OOPD OPGx-BEST1 Bestrophinopathies OPGx-RHO adRP co-funded by FFB &
NIH OPGx-RDH12 LCA co-funded by Global RDH12 Alliance OPGx-MERTK RP co-funded by FFB RD Fund & Abu Dhabi’s Healthcare Research and Innovation Fund OPGx-NMNAT1 LCA OPGx-CNGB1 RP NIH-funded consortium Undisclosed IRD GTx All gene
therapy programs have the potential to qualify for a Priority Review Voucher 9
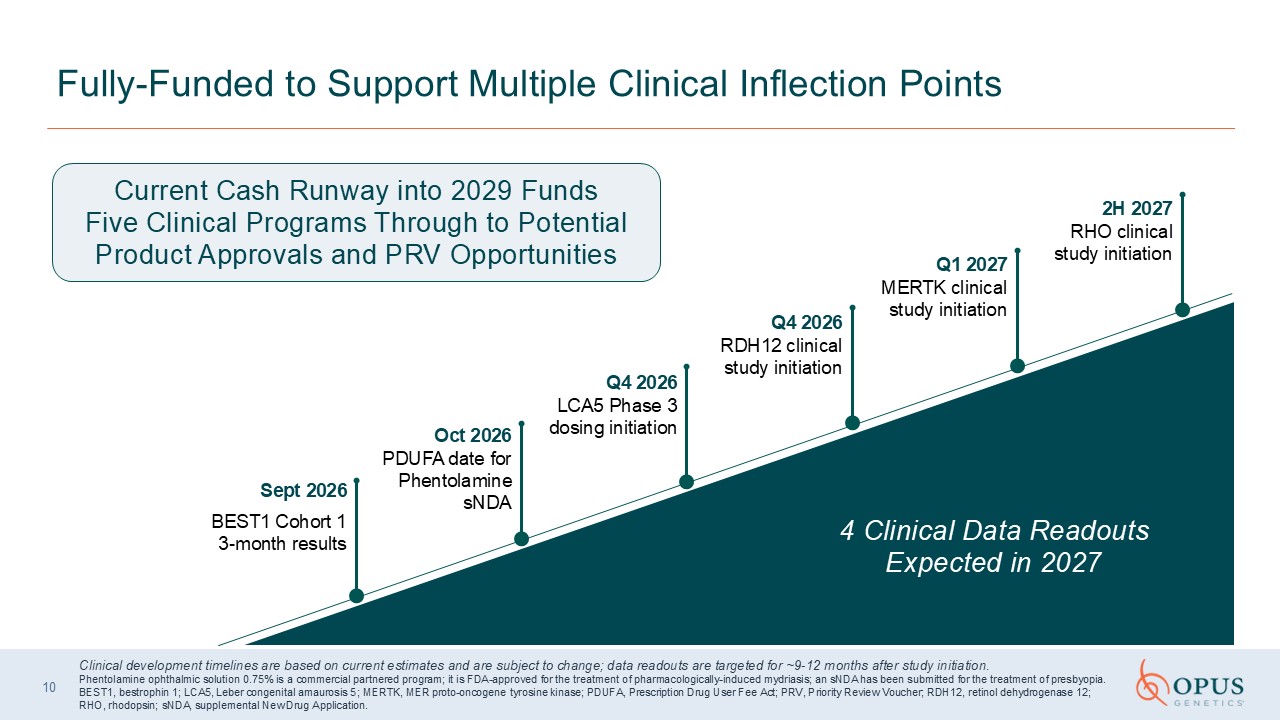
10 Fully-Funded to Support Multiple Clinical Inflection Points 2H 2027 RHO
clinical study initiation Q1 2027 MERTK clinical study initiation Q4 2026RDH12 clinical study initiation Q4 2026LCA5 Phase 3 dosing initiation Oct 2026PDUFA date for Phentolamine sNDA Sept 2026 BEST1 Cohort 1 3-month results Current
Cash Runway into 2029 Funds Five Clinical Programs Through to Potential Product Approvals and PRV Opportunities 4 Clinical Data Readouts Expected in 2027 Clinical development timelines are based on current estimates and are subject to
change; data readouts are targeted for ~9-12 months after study initiation. Phentolamine ophthalmic solution 0.75% is a commercial partnered program; it is FDA-approved for the treatment of pharmacologically-induced mydriasis; an sNDA has
been submitted for the treatment of presbyopia. BEST1, bestrophin 1; LCA5, Leber congenital amaurosis 5; MERTK, MER proto-oncogene tyrosine kinase; PDUFA, Prescription Drug User Fee Act; PRV, Priority Review Voucher; RDH12, retinol
dehydrogenase 12; RHO, rhodopsin; sNDA, supplemental New Drug Application.

Scientific OverviewJean Bennett, MD, PhDF.M. Kirby Emeritus Professor of
Ophthalmology Perelman School of Medicine, University of PennsylvaniaPhiladelphia, PA
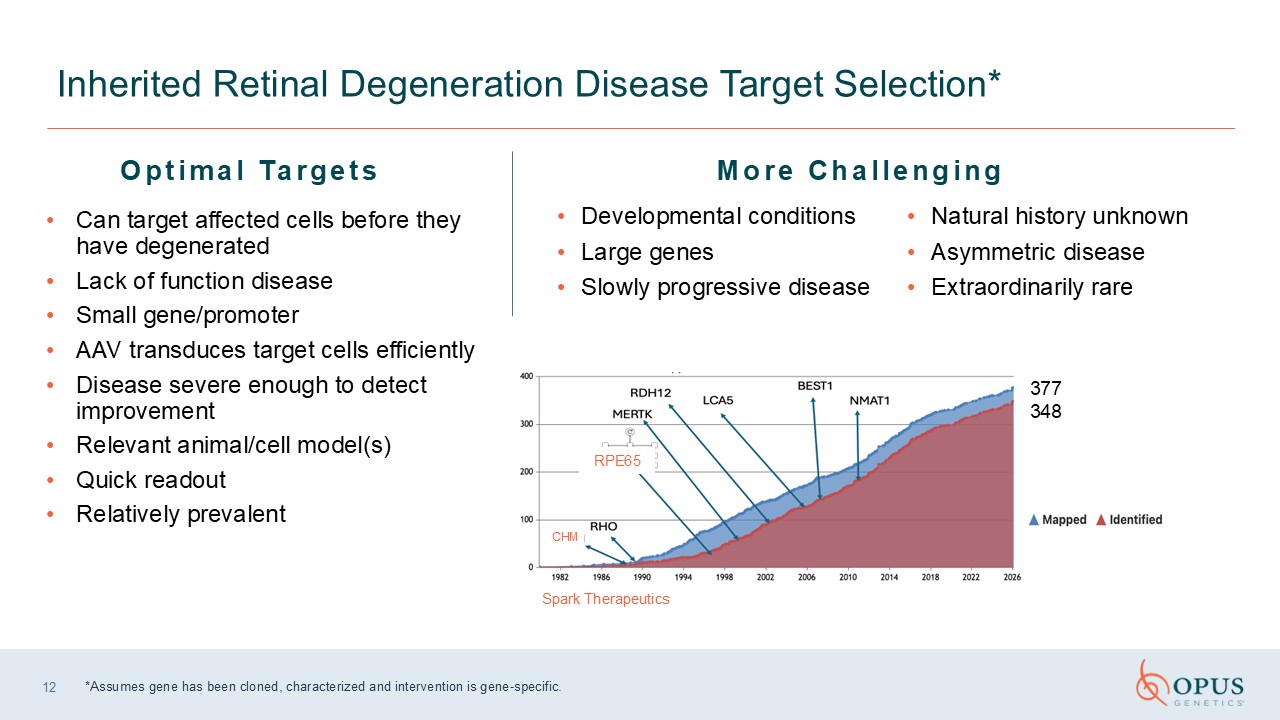
12 Inherited Retinal Degeneration Disease Target Selection* *Assumes gene has
been cloned, characterized and intervention is gene-specific. Optimal Targets Developmental conditions Large genes Slowly progressive disease Natural history unknown Asymmetric disease Extraordinarily rare Can target affected cells
before they have degenerated Lack of function disease Small gene/promoter AAV transduces target cells efficiently Disease severe enough to detect improvement Relevant animal/cell model(s) Quick readout Relatively prevalent More
Challenging Spark Therapeutics 377 348 RPE65 CHM

13 Treat the function to reverse pathology and restore or preserve
vision Structure-Function Dissociation: The Clinical Imperative Targeting Diseases Where the Structure is Intact Retinal structure is relatively preservedeven though visual function is already impaired This creates a “therapeutic window”
where there are still enough viable cells for AAV gene replacement to restore function Pick the right patients, and choose meaningful endpoints for our clinical trials Clinical evidence for curative potential in IRDs AAV, adeno-associated
virus; IRD, inherited retinal disease.

14 Inspiration from Patients and Families LCA-RDH12 Kids LCA-LCA5 and
LCA-RDH12: Ciliopathies Rare Imaging: Treatable photoreceptors in childhood, young adults Animal & cell models Compared to RPE65: More severe Earlier onset 1st to be identified with LCA5 LCA5, Leber congenital amaurosis 5;
RPE65, retinal pigment epithelium-specific 65 kDa protein; RDH12, retinol dehydrogenase 12.
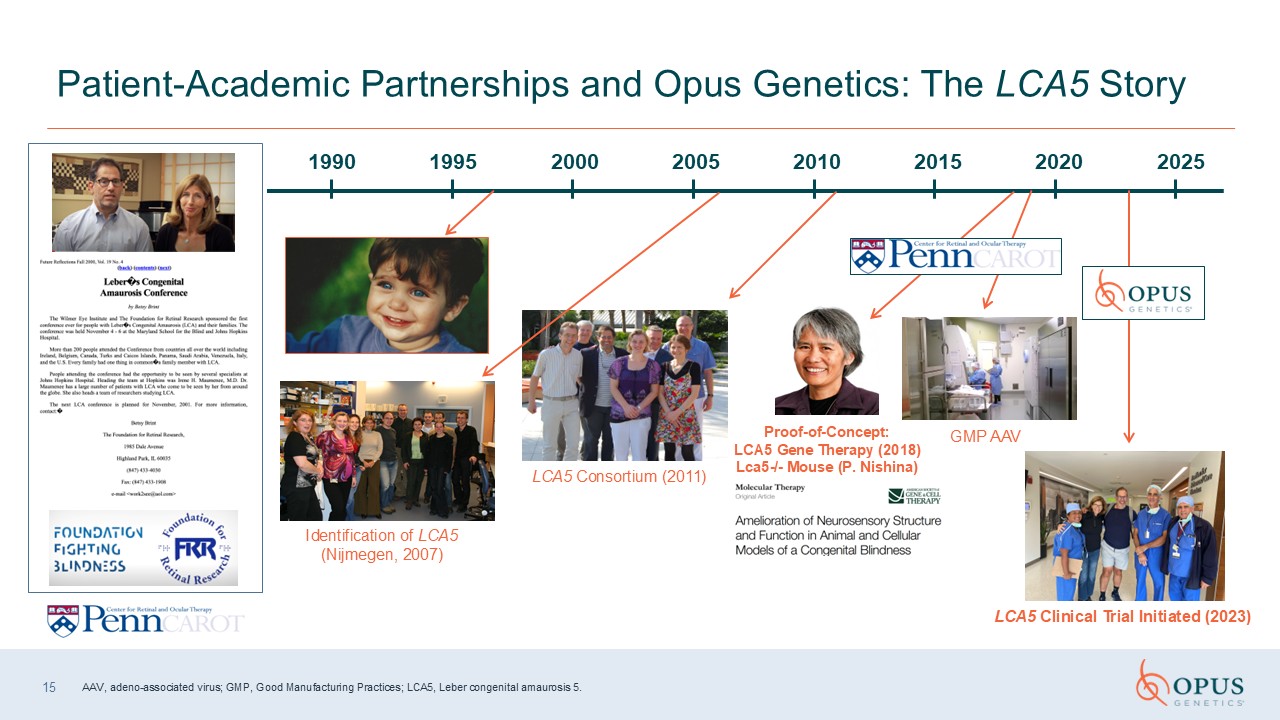
15 Patient-Academic Partnerships and Opus Genetics: The LCA5
Story Identification of LCA5 (Nijmegen, 2007) Proof-of-Concept: LCA5 Gene Therapy (2018) Lca5-/- Mouse (P. Nishina) LCA5 Consortium (2011) GMP AAV LCA5 Clinical Trial Initiated
(2023) 1990 1995 2000 2005 2025 2020 2015 2010 AAV, adeno-associated virus; GMP, Good Manufacturing Practices; LCA5, Leber congenital amaurosis 5.

Abundant Safety Data >140 retinal gene therapy clinical trials
initiated Gene therapy centers around the world Thousands of eyes have been injected Numerous disease targets Variety of strategies Excellent safety data Familiarity with gene therapy surgical procedures, vector handling and storage,
genotype-phenotype correlations, outcome measures Half a dozen retinal gene therapy clinical trials will read out within the next year Efficacy/Durability Data – LUXTURNA® (voretigene neparvovec-rzyl) Long-term durability data (>9 years
and counting) for Phase 3 Real world efficacy similar to that reported in clinical trials 16 Status of Retinal Gene Therapy LUXTURNA® is a registered trademark of Spark Therapeutics, Inc.
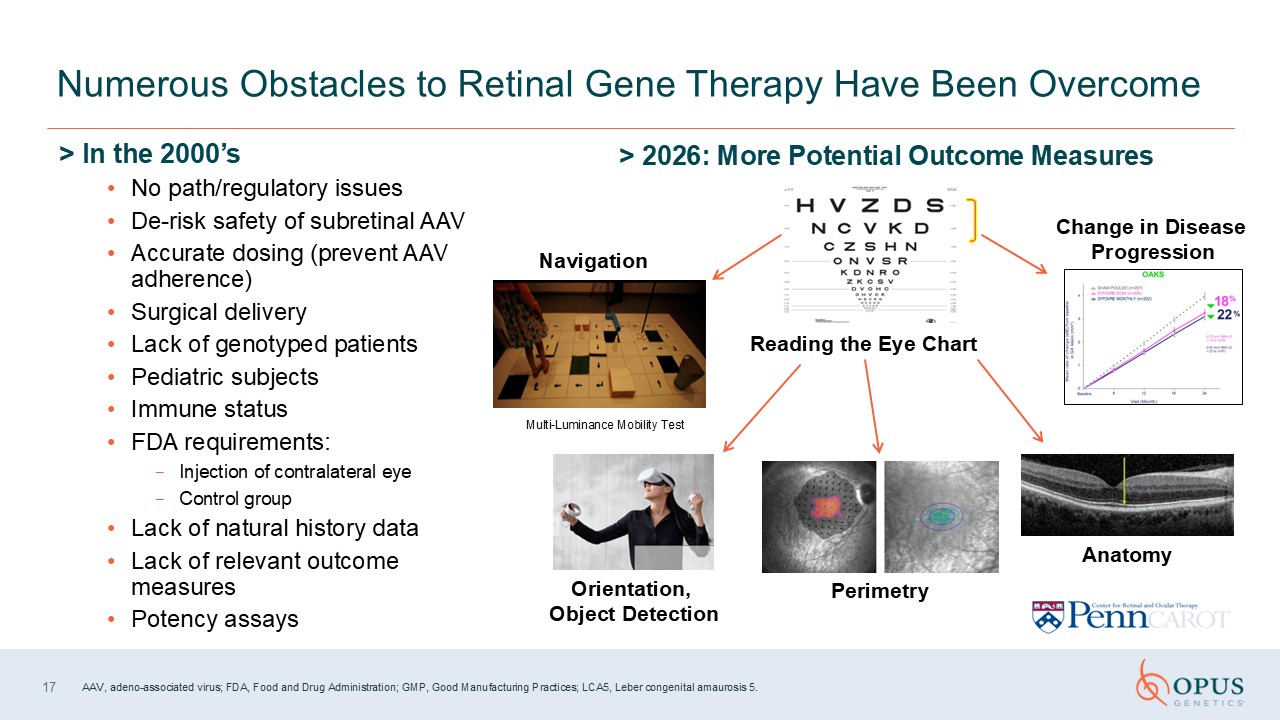
> In the 2000’s No path/regulatory issues De-risk safety of subretinal
AAV Accurate dosing (prevent AAV adherence) Surgical delivery Lack of genotyped patients Pediatric subjects Immune status FDA requirements: Injection of contralateral eye Control group Lack of natural history data Lack of relevant
outcome measures Potency assays Numerous Obstacles to Retinal Gene Therapy Have Been Overcome > 2026: More Potential Outcome Measures Multi-Luminance Mobility Test Navigation Orientation, Object
Detection Perimetry Anatomy Change in Disease Progression Reading the Eye Chart 17 AAV, adeno-associated virus; FDA, Food and Drug Administration; GMP, Good Manufacturing Practices; LCA5, Leber congenital amaurosis 5.

RDH12 Overview Kenneth Fan, MD, MBA Retina Consultants of Texas Houston, TX
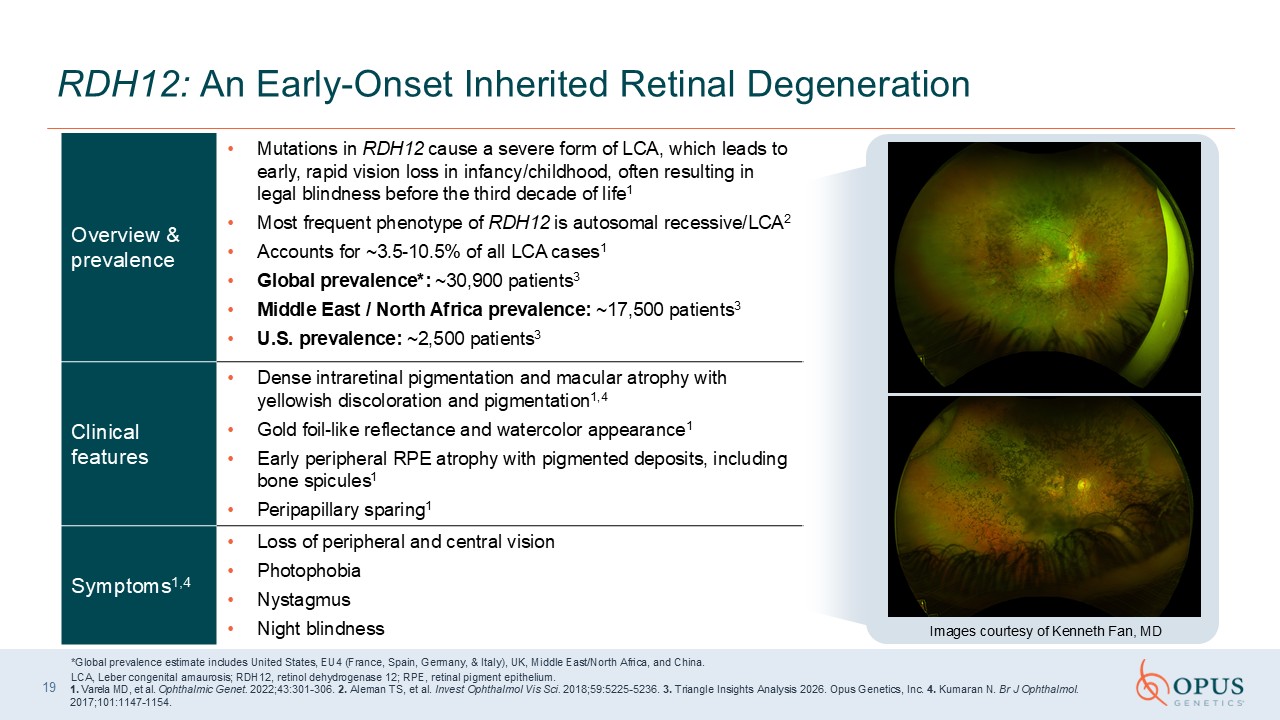
Overview & prevalence Mutations in RDH12 cause a severe form of LCA, which
leads to early, rapid vision loss in infancy/childhood, often resulting in legal blindness before the third decade of life1 Most frequent phenotype of RDH12 is autosomal recessive/LCA2 Accounts for ~3.5-10.5% of all LCA cases1 Global
prevalence*: ~30,900 patients3 Middle East / North Africa prevalence: ~17,500 patients3 U.S. prevalence: ~2,500 patients3 Clinical features Dense intraretinal pigmentation and macular atrophy with yellowish discoloration and
pigmentation1,4 Gold foil-like reflectance and watercolor appearance1 Early peripheral RPE atrophy with pigmented deposits, including bone spicules1 Peripapillary sparing1 Symptoms1,4 Loss of peripheral and central
vision Photophobia Nystagmus Night blindness 19 RDH12: An Early-Onset Inherited Retinal Degeneration *Global prevalence estimate includes United States, EU4 (France, Spain, Germany, & Italy), UK, Middle East/North Africa, and
China. LCA, Leber congenital amaurosis; RDH12, retinol dehydrogenase 12; RPE, retinal pigment epithelium. 1. Varela MD, et al. Ophthalmic Genet. 2022;43:301-306. 2. Aleman TS, et al. Invest Ophthalmol Vis Sci. 2018;59:5225-5236. 3. Triangle
Insights Analysis 2026. Opus Genetics, Inc. 4. Kumaran N. Br J Ophthalmol. 2017;101:1147-1154. Images courtesy of Kenneth Fan, MD
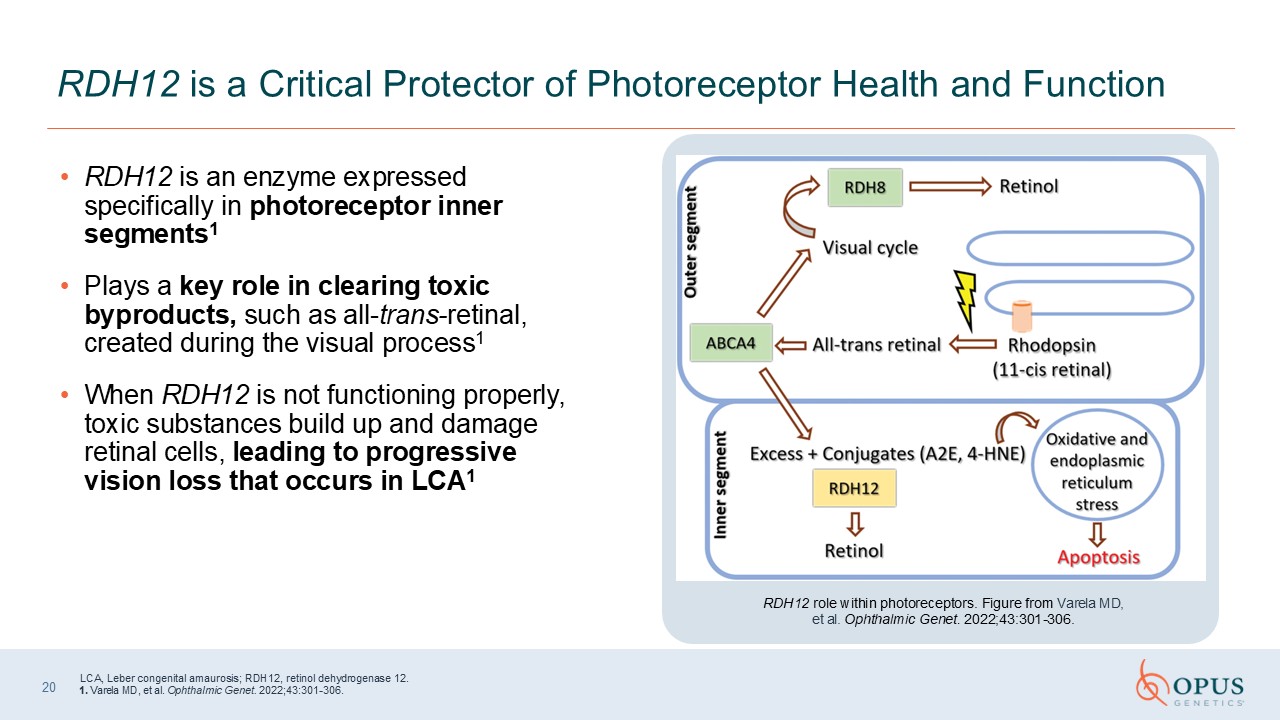
RDH12 is an enzyme expressed specifically in photoreceptor inner
segments1 Plays a key role in clearing toxic byproducts, such as all-trans-retinal, created during the visual process1 When RDH12 is not functioning properly, toxic substances build up and damage retinal cells, leading to progressive vision
loss that occurs in LCA1 RDH12 is a Critical Protector of Photoreceptor Health and Function RDH12 role within photoreceptors. Figure from Varela MD, et al. Ophthalmic Genet. 2022;43:301-306. 20 LCA, Leber congenital amaurosis; RDH12,
retinol dehydrogenase 12. 1. Varela MD, et al. Ophthalmic Genet. 2022;43:301-306.

Longitudinal studies of RDH12 patients suggest relatively preserved visual
acuity with steep decline in second decade of life Generally, VA maintained to ~20/200 or better for first two decades Thereafter, VA steeply declines toward finger counting, light perception only Functional signals remain in
extramacular rod function and in cone-mediated foveal vision Extramacular (peripheral) rod function is present even in face of VA decline Ideal treatment window is pre-adolescence, but patients up to 2nd decade may benefit prior to steep
VA decline 21 RDH12 Structural Changes Define the Therapeutic Window Structure-function dissociation creates favorable pathobiology for AAV gene replacement in RDH12 similar to RPE65 AAV, adeno-associated virus; ONL, outer nuclear layer;
ONL, outer nuclear layer; RDH12, retinol dehydrogenase 12; RPE65, retinal pigment epithelium-specific 65 kDa protein; VA, visual acuity. Jacobson SG, et al. Invest Ophthalmol Vis Sci. 2007;48:332–338 Generally lower ONL thickness, but
improved cone-mediated sensitivity in RDH12 patient (circled region indicates 1.5 log units or 30 times better sensitivity in RDH12 vs RPE65)

OPGx-RDH12 Scientific Overview Ash Jayagopal, PhD, MBA Chief Scientific and
Development Officer Opus Genetics
Success with LUXTURNA® indicates potential restoration of the retinoid cycle can
rescue retinal function Established AAV8 vector, codon optimized transgene, and specific promoter targets photoreceptors Designed to restore visual cycle function in the inner segments of photoreceptors OPGx-RDH12 Gene Therapy is Designed
to Restore a Key Component of the Visual Cycle OPGx-RDH12 Gene Therapy Vector AAV8 Delivery Single subretinal injection 23 AAV8, adeno-associated virus serotype 8; RK1, rhodopsin kinase 1.
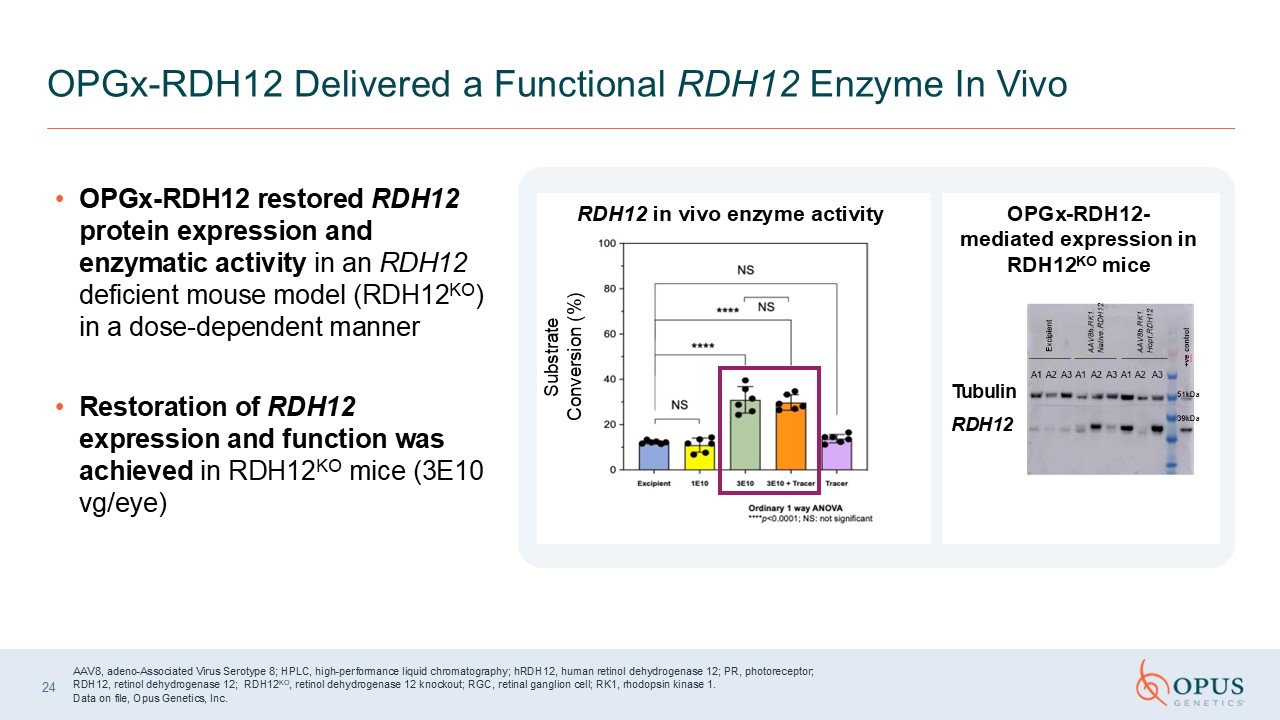
OPGx-RDH12 restored RDH12 protein expression and enzymatic activity in an RDH12
deficient mouse model (RDH12KO) in a dose-dependent manner Restoration of RDH12 expression and function was achieved in RDH12KO mice (3E10 vg/eye) OPGx-RDH12 Delivered a Functional RDH12 Enzyme In Vivo Substrate Conversion (%) RDH12 in
vivo enzyme activity OPGx-RDH12-mediated expression in RDH12KO mice RDH12 Tubulin AAV8, adeno-Associated Virus Serotype 8; HPLC, high-performance liquid chromatography; hRDH12, human retinol dehydrogenase 12; PR, photoreceptor; RDH12,
retinol dehydrogenase 12; RDH12KO, retinol dehydrogenase 12 knockout; RGC, retinal ganglion cell; RK1, rhodopsin kinase 1. Data on file, Opus Genetics, Inc. 24
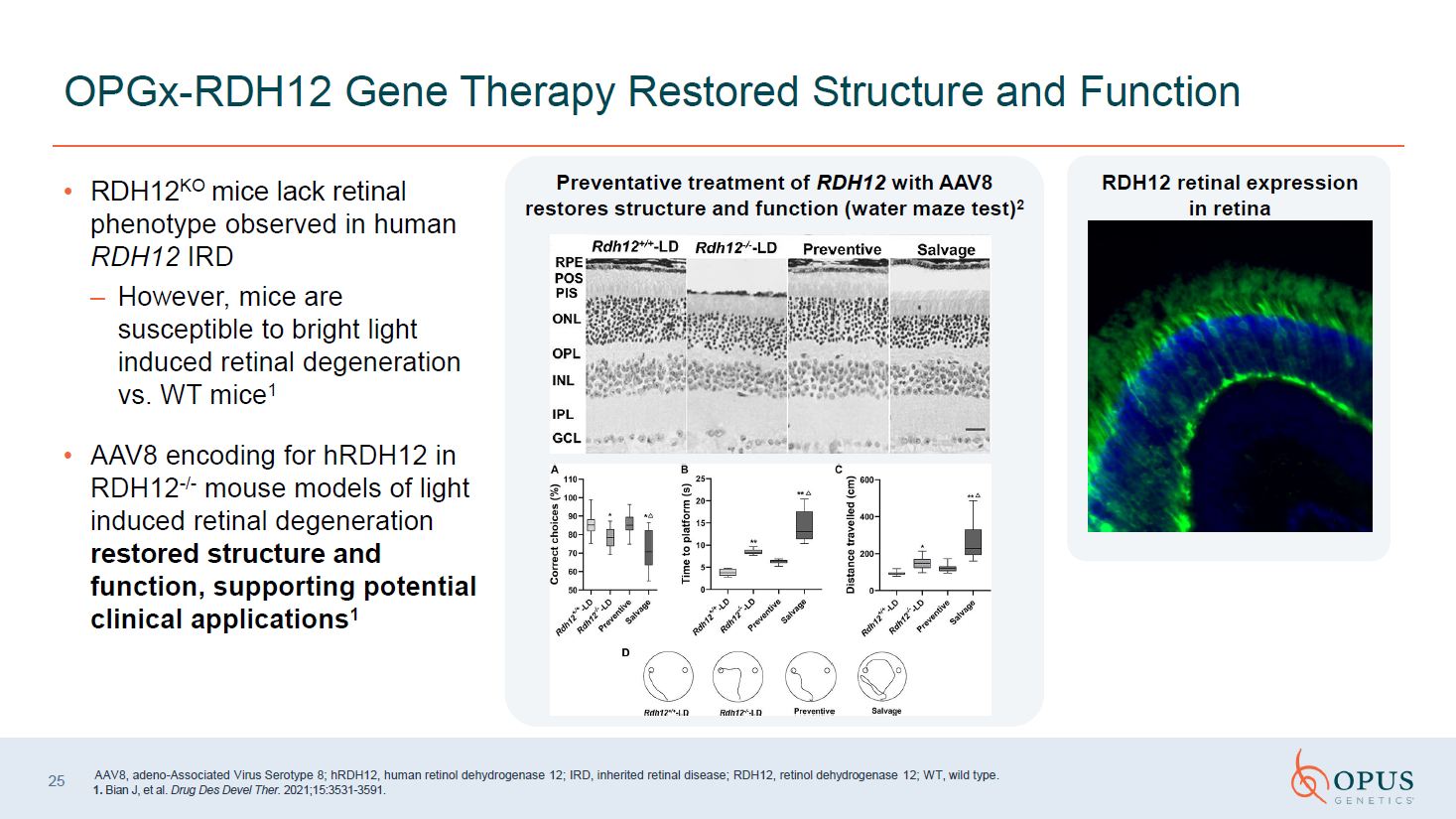
RDH12KO mice lack retinal phenotype
observed in human RDH12 IRD However, mice are susceptible to bright light induced retinal degeneration vs. WT mice1 AAV8 encoding for hRDH12 in RDH12-/- mouse models of light induced retinal degeneration restored structure and function,
supporting potential clinical applications1 OPGx-RDH12 Gene Therapy Restored Structure and Function Preventative treatment of RDH12 with AAV8 restores structure and function (water maze test)2 AAV8, adeno-Associated Virus Serotype 8;
hRDH12, human retinol dehydrogenase 12; IRD, inherited retinal disease; RDH12, retinol dehydrogenase 12; WT, wild type. 1. Bian J, et al. Drug Des Devel Ther. 2021;15:3531-3591.

MERTK Overview Professor Robert MacLarenUniversity of Oxford Oxford, England
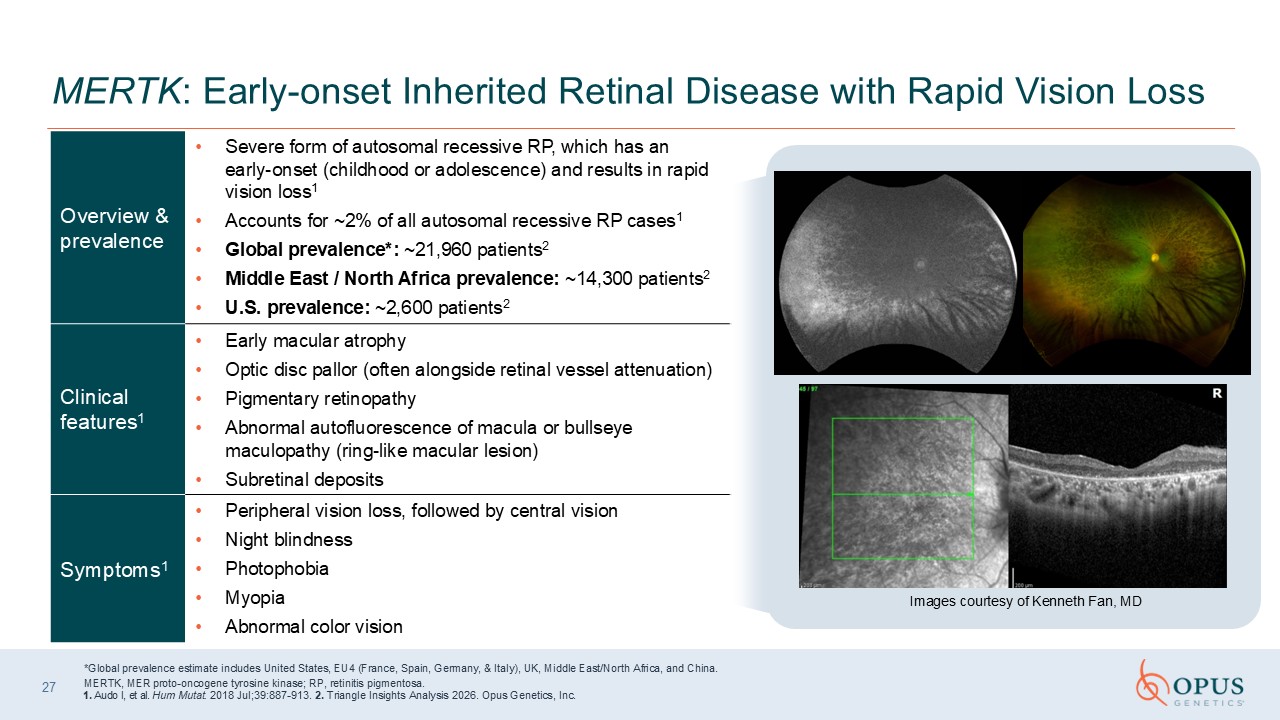
MERTK: Early-onset Inherited Retinal Disease with Rapid Vision Loss *Global
prevalence estimate includes United States, EU4 (France, Spain, Germany, & Italy), UK, Middle East/North Africa, and China. MERTK, MER proto-oncogene tyrosine kinase; RP, retinitis pigmentosa. 1. Audo I, et al. Hum Mutat. 2018
Jul;39:887-913. 2. Triangle Insights Analysis 2026. Opus Genetics, Inc. Images courtesy of Kenneth Fan, MD 27 Overview & prevalence Severe form of autosomal recessive RP, which has an early-onset (childhood or adolescence) and results
in rapid vision loss1 Accounts for ~2% of all autosomal recessive RP cases1 Global prevalence*: ~21,960 patients2 Middle East / North Africa prevalence: ~14,300 patients2 U.S. prevalence: ~2,600 patients2 Clinical features1 Early
macular atrophy Optic disc pallor (often alongside retinal vessel attenuation) Pigmentary retinopathy Abnormal autofluorescence of macula or bullseye maculopathy (ring-like macular lesion) Subretinal deposits Symptoms1 Peripheral vision
loss, followed by central vision Night blindness Photophobia Myopia Abnormal color vision
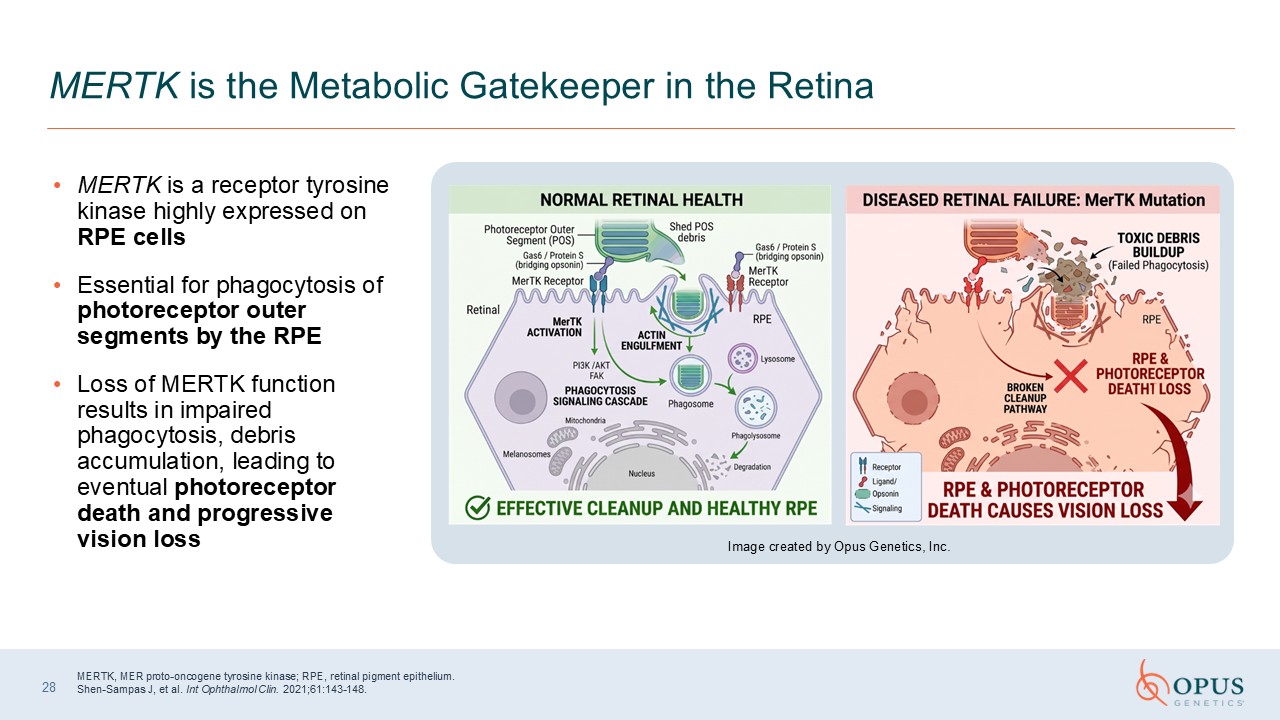
MERTK is a receptor tyrosine kinase highly expressed on RPE cells Essential for
phagocytosis of photoreceptor outer segments by the RPE Loss of MERTK function results in impaired phagocytosis, debris accumulation, leading to eventual photoreceptor death and progressive vision loss 28 MERTK is the Metabolic Gatekeeper
in the Retina MERTK, MER proto-oncogene tyrosine kinase; RPE, retinal pigment epithelium. Shen-Sampas J, et al. Int Ophthalmol Clin. 2021;61:143-148. Image created by Opus Genetics, Inc.
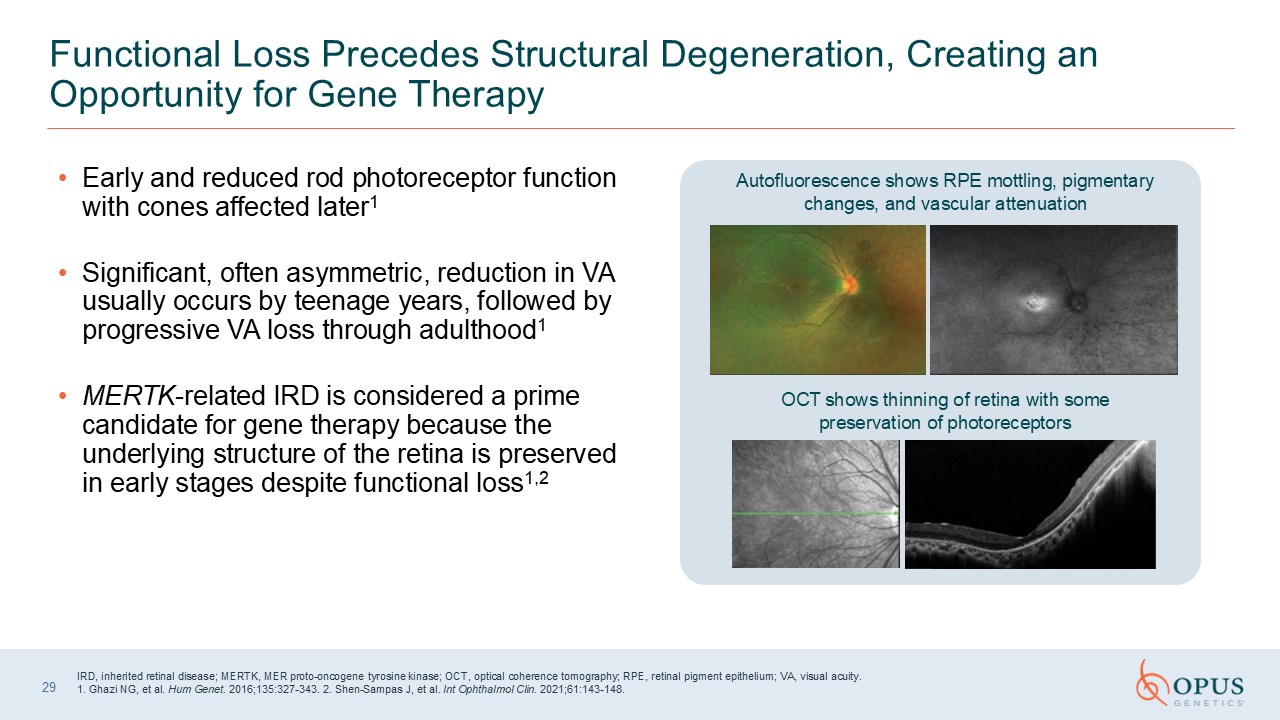
Early and reduced rod photoreceptor function with cones affected
later1 Significant, often asymmetric, reduction in VA usually occurs by teenage years, followed by progressive VA loss through adulthood1 MERTK-related IRD is considered a prime candidate for gene therapy because the underlying structure of
the retina is preserved in early stages despite functional loss1,2 29 Functional Loss Precedes Structural Degeneration, Creating an Opportunity for Gene Therapy IRD, inherited retinal disease; MERTK, MER proto-oncogene tyrosine kinase;
OCT, optical coherence tomography; RPE, retinal pigment epithelium; VA, visual acuity. 1. Ghazi NG, et al. Hum Genet. 2016;135:327-343. 2. Shen-Sampas J, et al. Int Ophthalmol Clin. 2021;61:143-148. Autofluorescence shows RPE mottling,
pigmentary changes, and vascular attenuation OCT shows thinning of retina with some preservation of photoreceptors

OPGx-MERTK Scientific Overview Ash Jayagopal, PhD, MBA Chief Scientific and
Development Officer Opus Genetics

Vector AAV2 Delivery Single subretinal injection Same AAV2 capsid used in
LUXTURNA® OPGx-MERTK is designed to restore critical RPE metabolic functions needed for retinal homeostasis RPE-specificity conferred by promoter technology 31 OPGx-MERTK Gene Therapy is Designed to Restore Critical Retinal Pigment
Epithelium Metabolic Functions AAV2, adeno-associated virus serotype 8; ITR, inverted terminal repeat; RPE, retinal pigment epithelium; VMD2, vitelliform macular dystrophy 2. OPGx-MERTK Gene Therapy
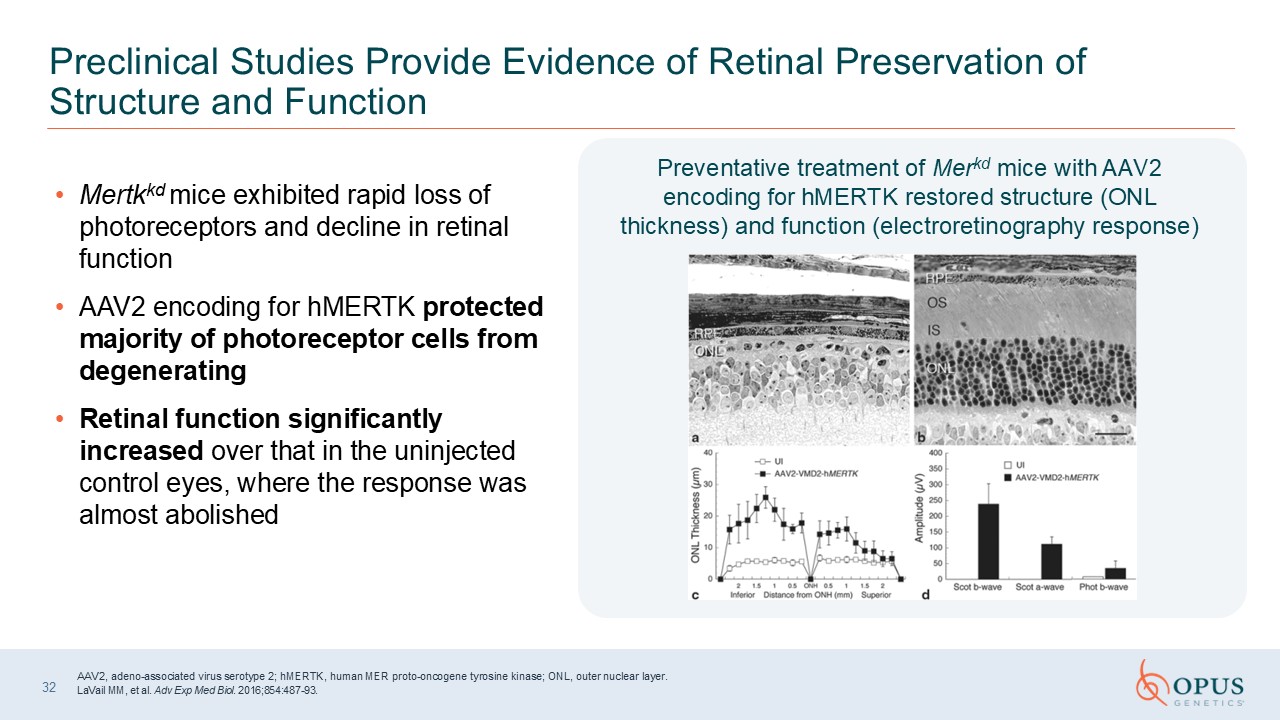
32 Preclinical Studies Provide Evidence of Retinal Preservation of Structure
and Function AAV2, adeno-associated virus serotype 2; hMERTK, human MER proto-oncogene tyrosine kinase; ONL, outer nuclear layer. LaVail MM, et al. Adv Exp Med Biol. 2016;854:487-93. Mertkkd mice exhibited rapid loss of photoreceptors
and decline in retinal function AAV2 encoding for hMERTK protected majority of photoreceptor cells from degenerating Retinal function significantly increased over that in the uninjected control eyes, where the response was almost
abolished Preventative treatment of Merkd mice with AAV2 encoding for hMERTK restored structure (ONL thickness) and function (electroretinography response)
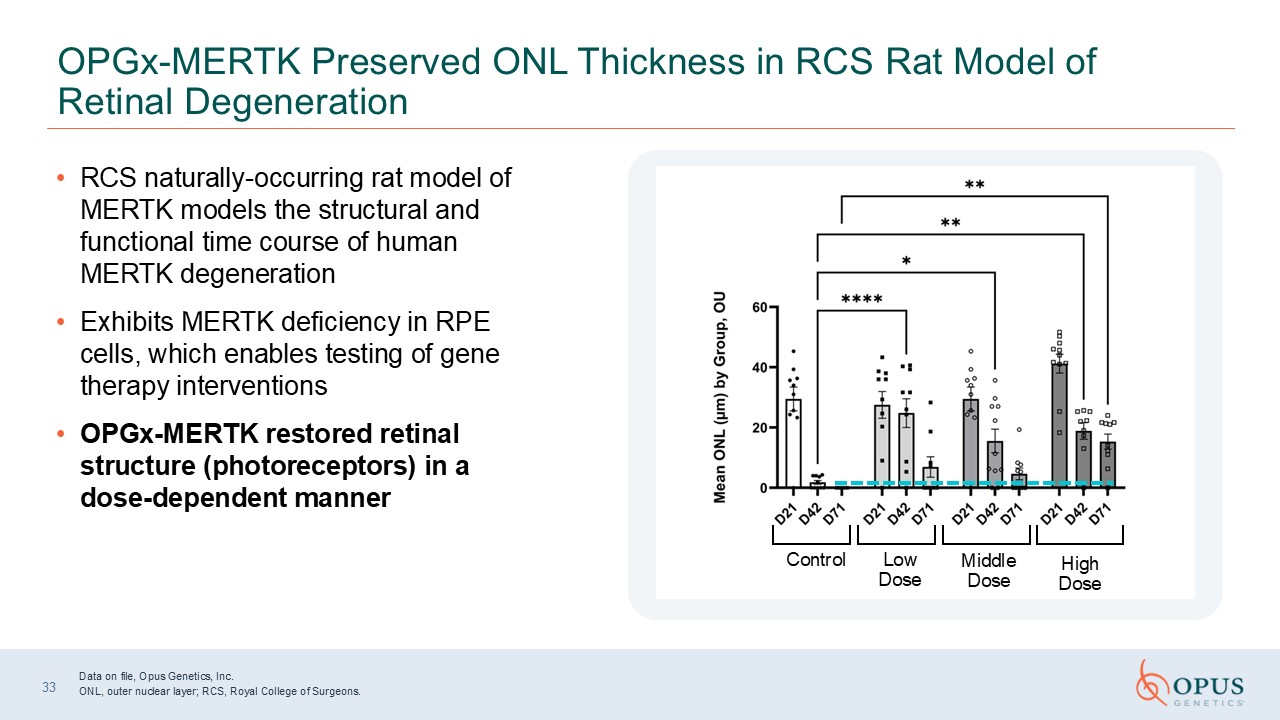
33 OPGx-MERTK Preserved ONL Thickness in RCS Rat Model of Retinal
Degeneration RCS naturally-occurring rat model of MERTK models the structural and functional time course of human MERTK degeneration Exhibits MERTK deficiency in RPE cells, which enables testing of gene therapy interventions OPGx-MERTK
restored retinal structure (photoreceptors) in a dose-dependent manner Control Low Dose Middle Dose High Dose Data on file, Opus Genetics, Inc. ONL, outer nuclear layer; RCS, Royal College of Surgeons.

RHO Overview Lejla Vajzovic, MD Duke University Durham, NC
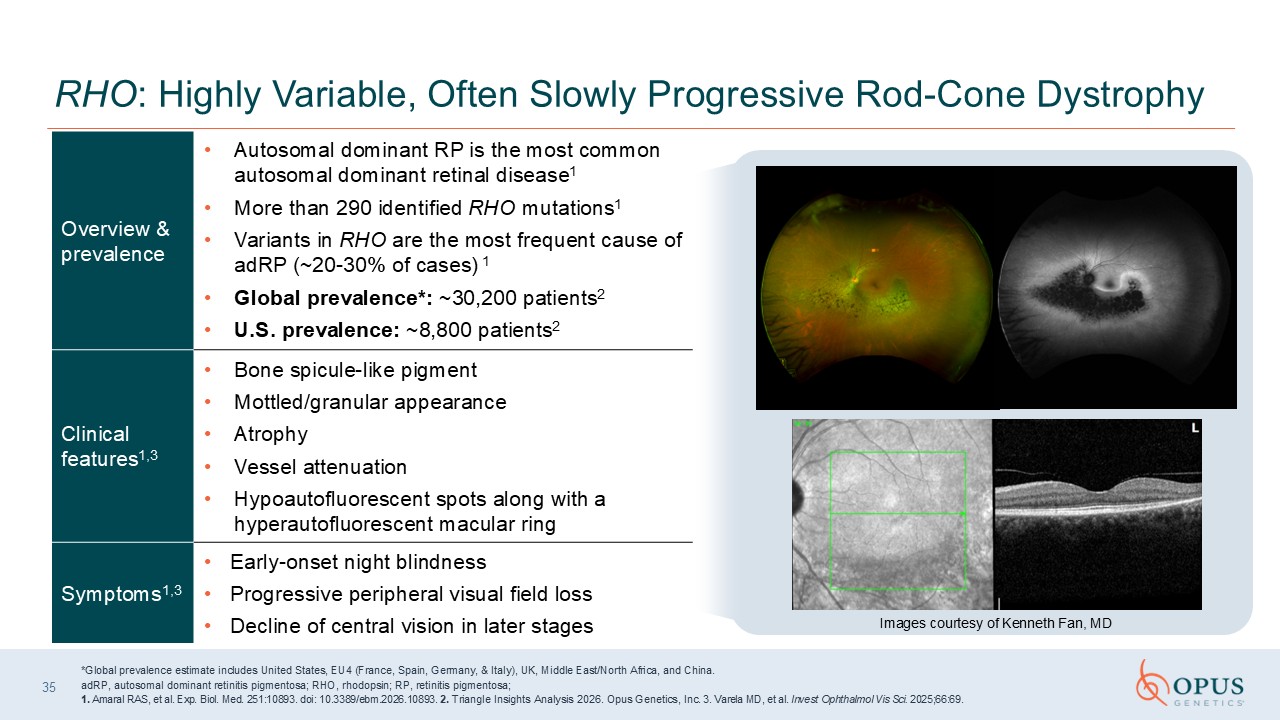
Overview & prevalence Autosomal dominant RP is the most common autosomal
dominant retinal disease1 More than 290 identified RHO mutations1 Variants in RHO are the most frequent cause of adRP (~20-30% of cases) 1 Global prevalence*: ~30,200 patients2 U.S. prevalence: ~8,800 patients2 Clinical features1,3 Bone
spicule-like pigment Mottled/granular appearance Atrophy Vessel attenuation Hypoautofluorescent spots along with a hyperautofluorescent macular ring Symptoms1,3 Early-onset night blindness Progressive peripheral visual field
loss Decline of central vision in later stages 35 RHO: Highly Variable, Often Slowly Progressive Rod-Cone Dystrophy 6/15/2026 *Global prevalence estimate includes United States, EU4 (France, Spain, Germany, & Italy), UK, Middle
East/North Africa, and China. adRP, autosomal dominant retinitis pigmentosa; RHO, rhodopsin; RP, retinitis pigmentosa; 1. Amaral RAS, et al. Exp. Biol. Med. 251:10893. doi: 10.3389/ebm.2026.10893. 2. Triangle Insights Analysis 2026. Opus
Genetics, Inc. 3. Varela MD, et al. Invest Ophthalmol Vis Sci. 2025;66:69. Images courtesy of Kenneth Fan, MD

Misfolded or Dysfunctional Rhodopsin Drives Photoreceptor Stress and Progressive
Rod-Cone Degeneration RHO encodes for rhodopsin, a rod photoreceptor 11-cis-retinaldehyde photopigment located in the outer segment, critical for phototransduction Mutations in RHO can cause the rhodopsin protein to misfold or function
abnormally, leading to progressive photoreceptor degeneration and vision loss seen in adRP Dominant RHO mutations are typically gain-of-function, necessitating ablate (knockdown) and replace gene therapy strategies, or dominant-negative,
which affects wild type rhodopsin protein function 36 adRP, autosomal dominant retinitis pigmentosa; RHO, rhodopsin. Athanasiou et al. Prog Retin Eye Res. 2018;62:1-23. 36 Schematic illustration of the potential pathogenic consequences
caused by rhodopsin mutations
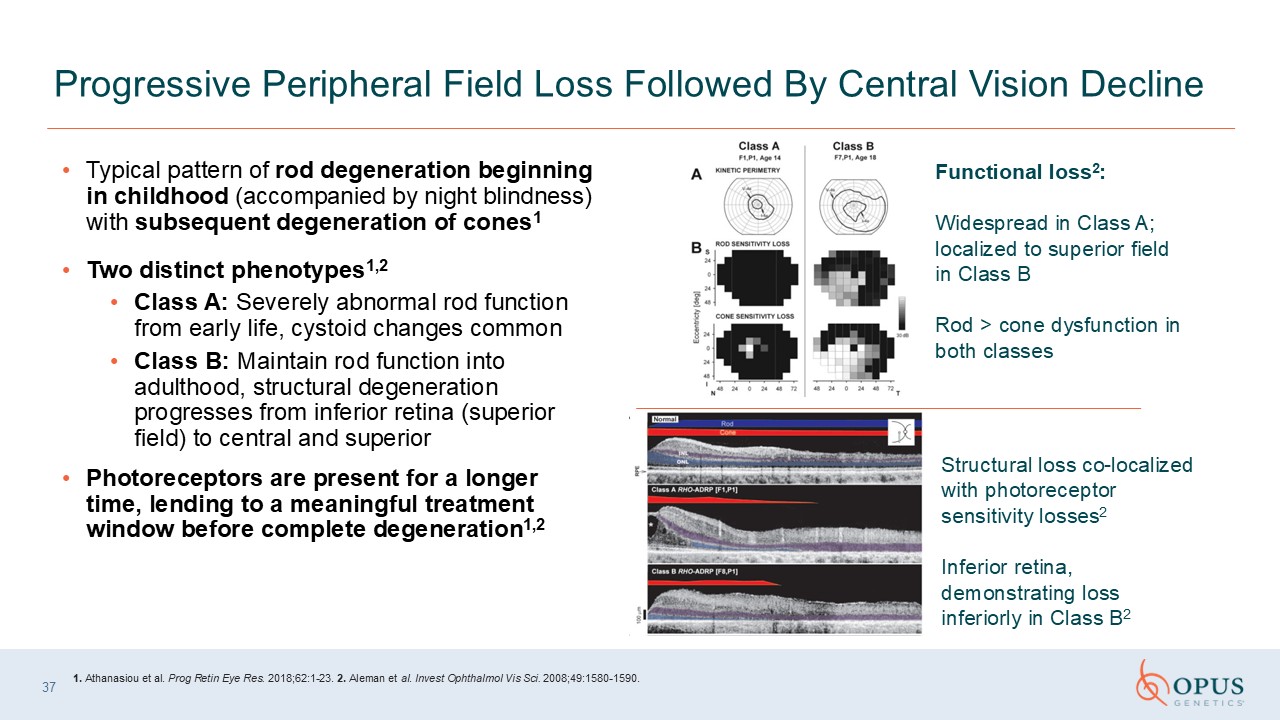
Typical pattern of rod degeneration beginning in childhood (accompanied by night
blindness) with subsequent degeneration of cones1 Two distinct phenotypes1,2 Class A: Severely abnormal rod function from early life, cystoid changes common Class B: Maintain rod function into adulthood, structural degeneration progresses
from inferior retina (superior field) to central and superior Photoreceptors are present for a longer time, lending to a meaningful treatment window before complete degeneration1,2 Progressive Peripheral Field Loss Followed By Central
Vision Decline Functional loss2: Widespread in Class A; localized to superior field in Class B Rod > cone dysfunction in both classes Structural loss co-localized with photoreceptor sensitivity losses2 Inferior retina,
demonstrating loss inferiorly in Class B2 1. Athanasiou et al. Prog Retin Eye Res. 2018;62:1-23. 2. Aleman et al. Invest Ophthalmol Vis Sci. 2008;49:1580-1590. 37

OPGx-RHO Scientific Overview Ash Jayagopal, PhD, MBA Chief Scientific and
Development Officer Opus Genetics

Vector AAV5 Delivery Single subretinal injection 39 OPGx-RHO Gene Therapy
is Designed to “Silence and Replace” in Autosomal Dominant RHO Silence (shRNA820) and replace (RHO820) strategy Use of AAV5 and rod-specific promoter Targets expression of codon-optimized human RHO transgene to rod and cone photoreceptors
while selectively suppressing the endogenous mutant (and toxic) RHO protein AAV, adeno-associated virus; RHO, rhodopsin, scAAV5, self-complementary adeno-associated virus serotype 5; WT, wild type. OPGx-RHO Gene Therapy
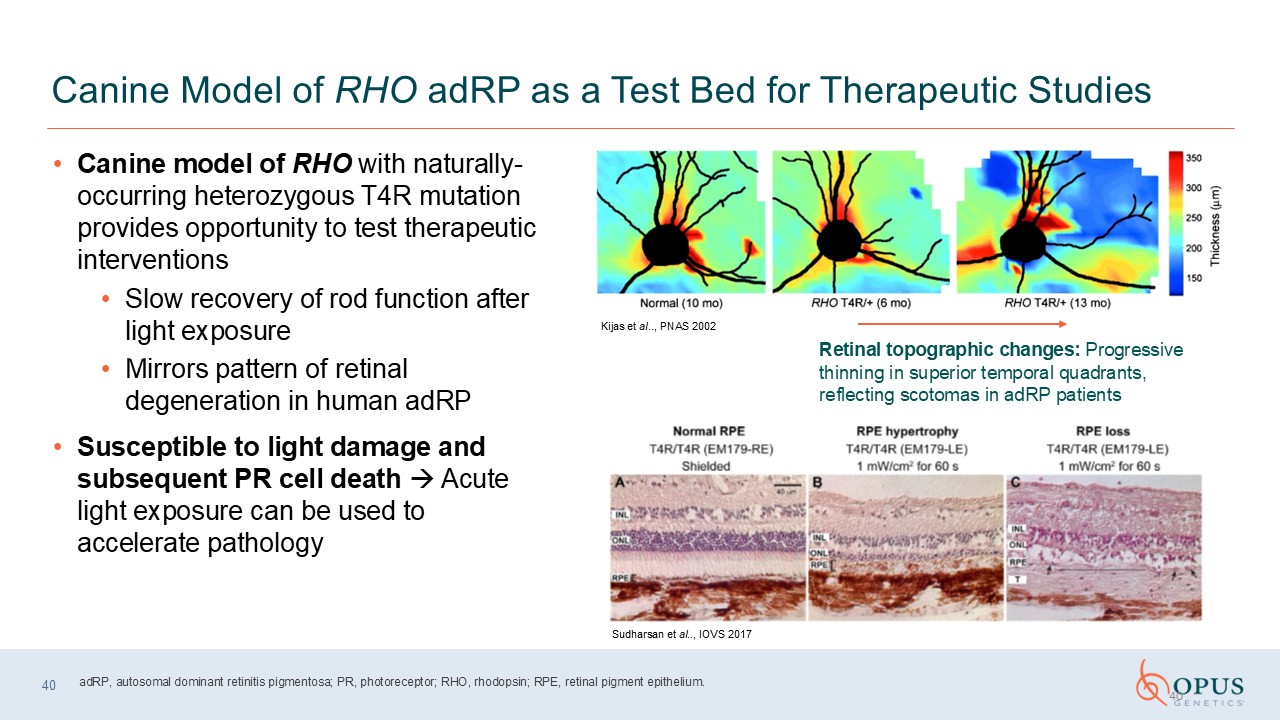
Canine Model of RHO adRP as a Test Bed for Therapeutic Studies Canine model of
RHO with naturally-occurring heterozygous T4R mutation provides opportunity to test therapeutic interventions Slow recovery of rod function after light exposure Mirrors pattern of retinal degeneration in human adRP Susceptible to light
damage and subsequent PR cell death Acute light exposure can be used to accelerate pathology 40 Retinal topographic changes: Progressive thinning in superior temporal quadrants, reflecting scotomas in adRP patients Kijas et al.., PNAS
2002 Sudharsan et al.., IOVS 2017 adRP, autosomal dominant retinitis pigmentosa; PR, photoreceptor; RHO, rhodopsin; RPE, retinal pigment epithelium. 40
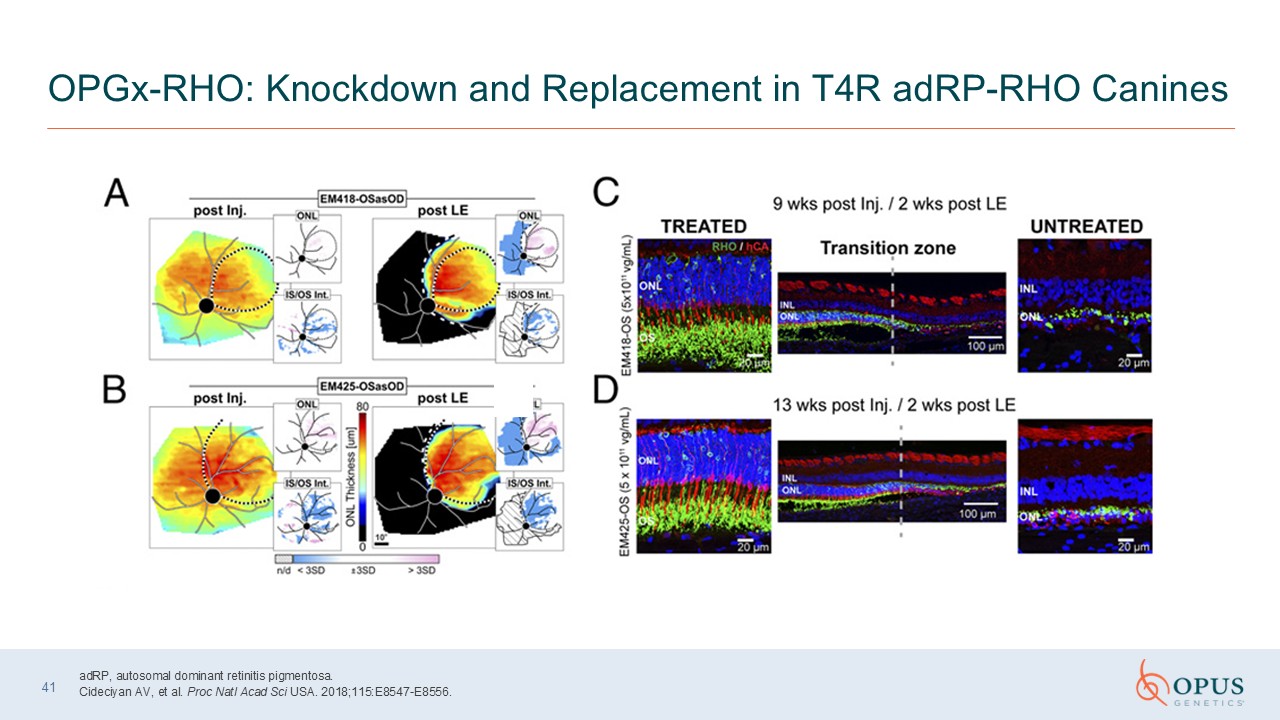
41 OPGx-RHO: Knockdown and Replacement in T4R adRP-RHO Canines adRP, autosomal
dominant retinitis pigmentosa. Cideciyan AV, et al. Proc Natl Acad Sci USA. 2018;115:E8547-E8556.
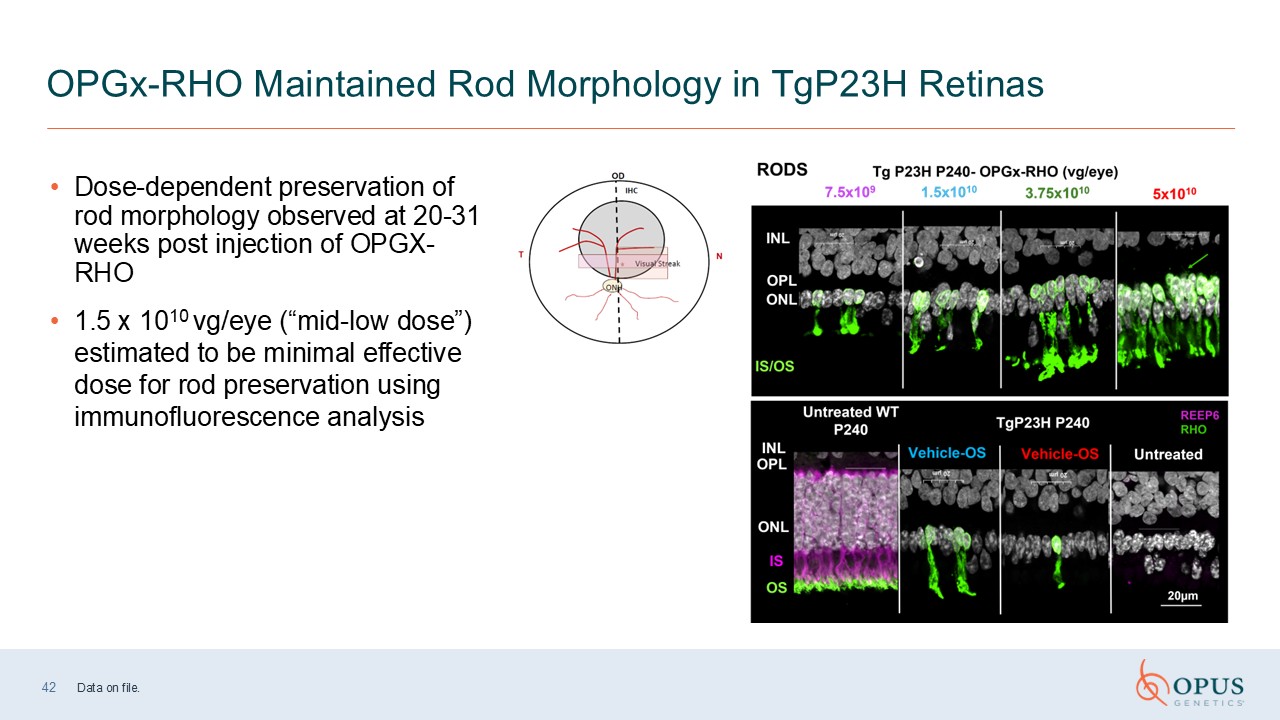
42 OPGx-RHO Maintained Rod Morphology in TgP23H Retinas Dose-dependent
preservation of rod morphology observed at 20-31 weeks post injection of OPGX-RHO 1.5 x 1010 vg/eye (“mid-low dose”) estimated to be minimal effective dose for rod preservation using immunofluorescence analysis Data on file.
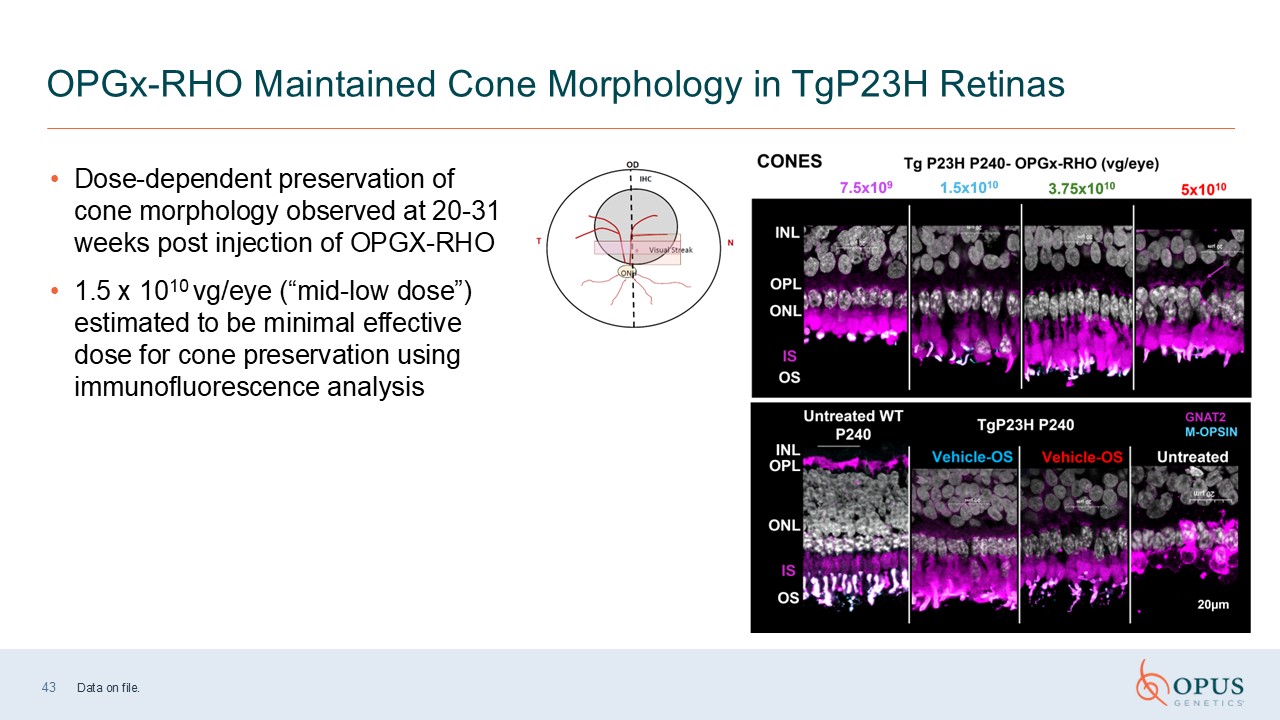
43 OPGx-RHO Maintained Cone Morphology in TgP23H Retinas Dose-dependent
preservation of cone morphology observed at 20-31 weeks post injection of OPGX-RHO 1.5 x 1010 vg/eye (“mid-low dose”) estimated to be minimal effective dose for cone preservation using immunofluorescence analysis Data on file.

Clinical Development Strategy Sally Tucker, PhD Chief Medical Officer Opus
Genetics
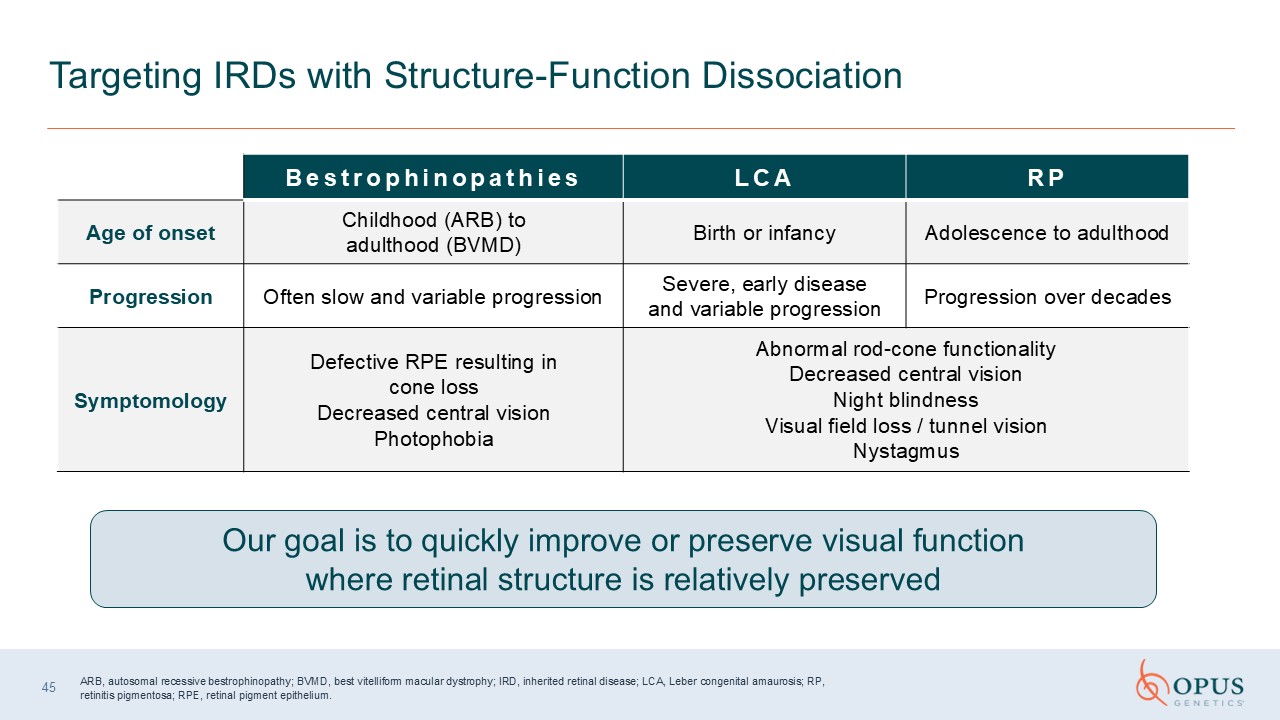
45 Targeting IRDs with Structure-Function
Dissociation Bestrophinopathies LCA RP Age of onset Childhood (ARB) to adulthood (BVMD) Birth or infancy Adolescence to adulthood Progression Often slow and variable progression Severe, early disease and variable
progression Progression over decades Symptomology Defective RPE resulting in cone loss Decreased central vision Photophobia Abnormal rod-cone functionality Decreased central vision Night blindness Visual field loss / tunnel
vision Nystagmus Our goal is to quickly improve or preserve visual function where retinal structure is relatively preserved ARB, autosomal recessive bestrophinopathy; BVMD, best vitelliform macular dystrophy; IRD, inherited retinal
disease; LCA, Leber congenital amaurosis; RP, retinitis pigmentosa; RPE, retinal pigment epithelium.
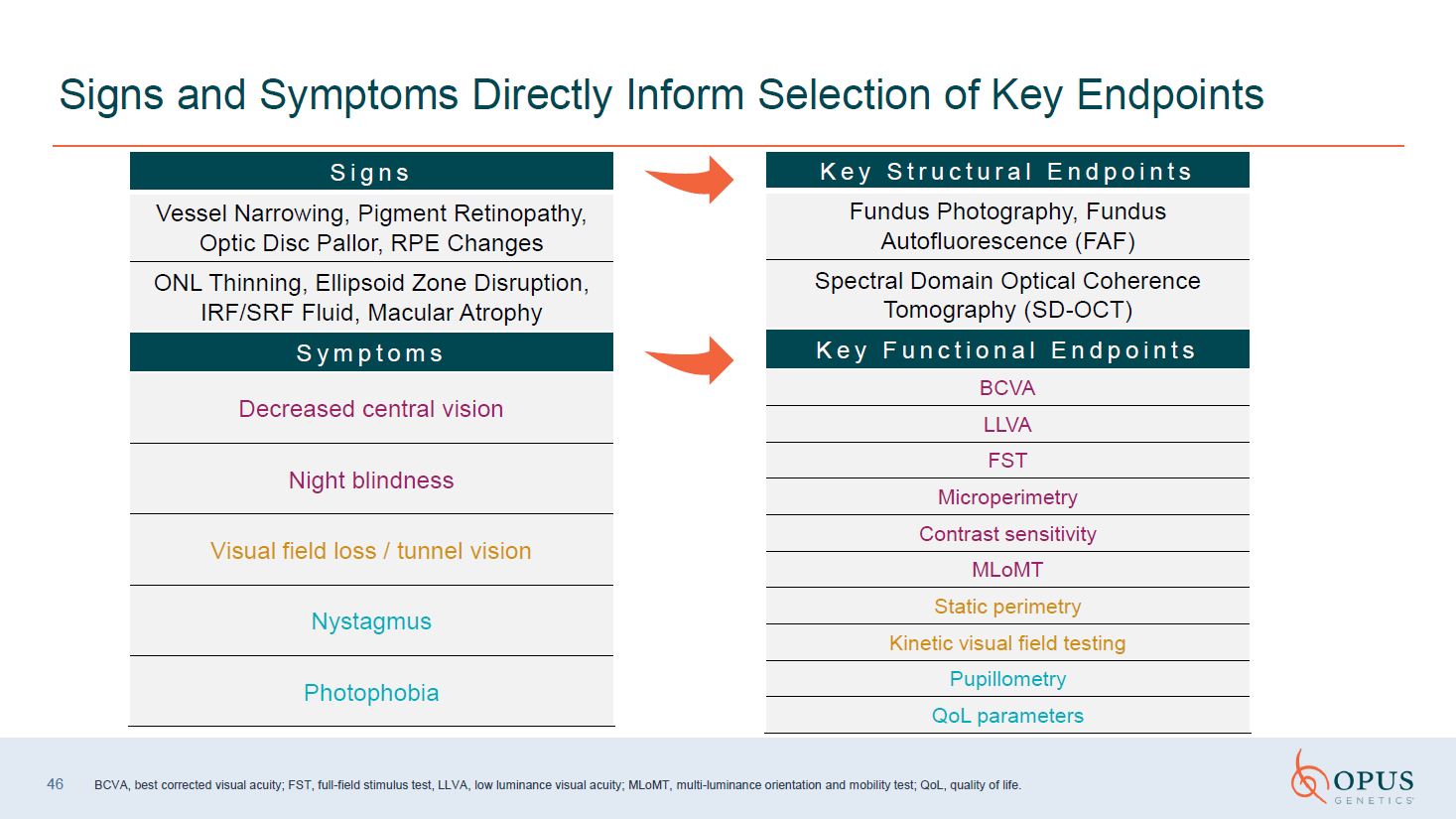
46 Signs and Symptoms Directly Inform Selection of Key
Endpoints Signs Vessel Narrowing, Pigment Retinopathy, Optic Disc Pallor, RPE Changes ONL Thinning, Ellipsoid Zone Disruption, IRF/SRF Fluid, Macular Atrophy Symptoms Decreased central vision Night blindness Visual field loss / tunnel
vision Nystagmus Photophobia Key Structural Endpoints Fundus Photography, Fundus Autofluorescence (FAF) Spectral Domain Optical Coherence Tomography (SD-OCT) Key Functional Endpoints BCVA LLVA FST Microperimetry Contrast
sensitivity MLoMT Static perimetry Kinetic visual field testing Pupillometry QoL parameters BCVA, best corrected visual acuity; FST, full-field stimulus test, MLoMT, multi-luminance orientation and mobility test; QoL, quality of life.
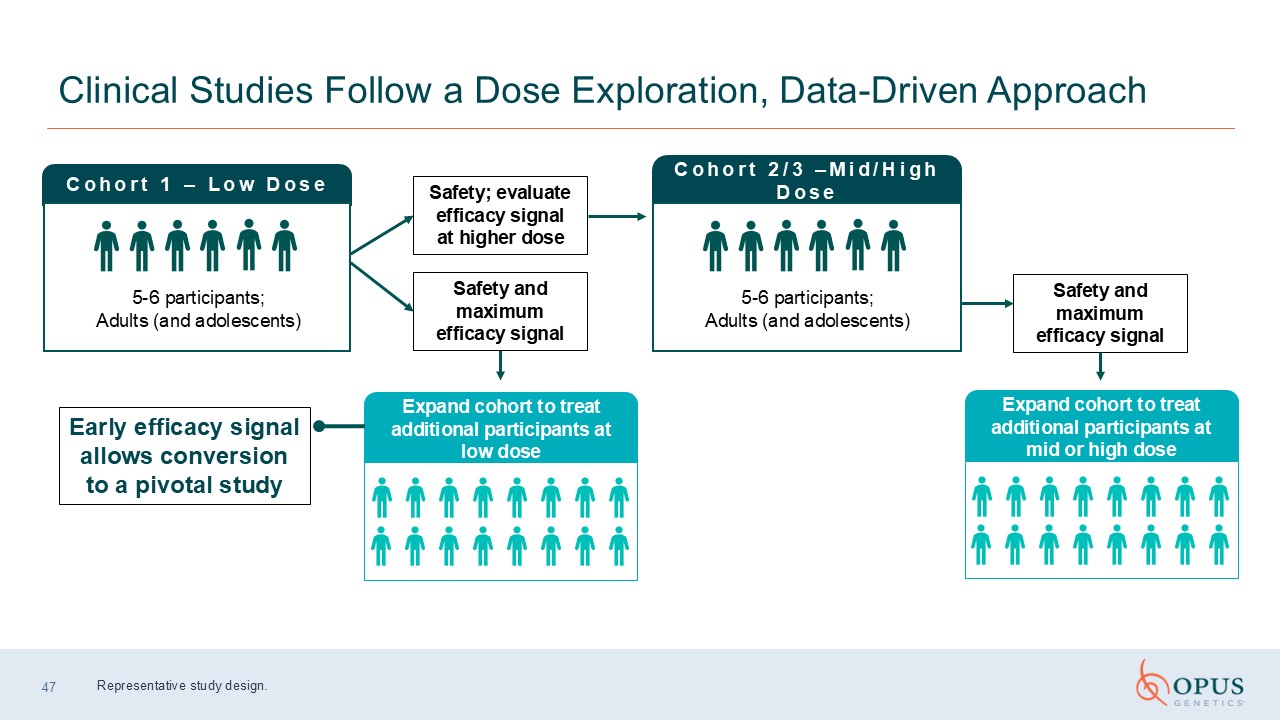
47 Clinical Studies Follow a Dose Exploration, Data-Driven Approach Cohort 1 –
Low Dose 5-6 participants; Adults (and adolescents) Early efficacy signal allows conversion to a pivotal study Safety; evaluate efficacy signal at higher dose Cohort 2/3 –Mid/High Dose 5-6 participants; Adults (and adolescents) Safety
and maximum efficacy signal Expand cohort to treat additional participants at low dose Safety and maximum efficacy signal Representative study design. Expand cohort to treat additional participants at mid or high dose

48 Targeting Initiation of Clinical Testing in All Three Programs within the
Next Year Program Region Study Initiation RDH12 U.S. Q4 2026 MERTK Abu Dhabi Q1 2027 RHO Global locations 2H 2027 Clinical development timelines are based on current estimates and are subject to change. MERTK, MER proto-oncogene
tyrosine kinase; RDH12, retinol dehydrogenase 12; RHO, rhodopsin.
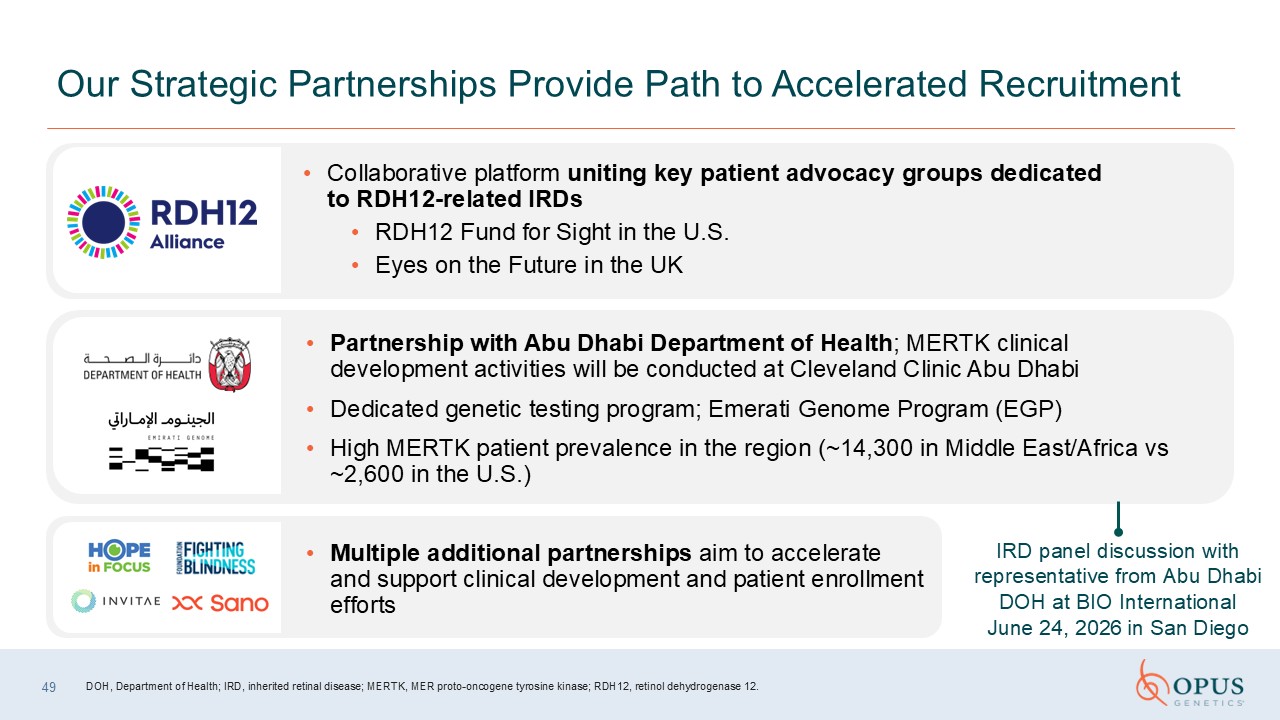
49 Our Strategic Partnerships Provide Path to Accelerated
Recruitment Collaborative platform uniting key patient advocacy groups dedicated to RDH12-related IRDs RDH12 Fund for Sight in the U.S. Eyes on the Future in the UK Partnership with Abu Dhabi Department of Health; MERTK clinical
development activities will be conducted at Cleveland Clinic Abu Dhabi Dedicated genetic testing program; Emerati Genome Program (EGP) High MERTK patient prevalence in the region (~14,300 in Middle East/Africa vs ~2,600 in the
U.S.) Multiple additional partnerships aim to accelerate and support clinical development and patient enrollment efforts IRD panel discussion with representative from Abu Dhabi DOH at BIO International June 24, 2026 in San Diego DOH,
Department of Health; IRD, inherited retinal disease; MERTK, MER proto-oncogene tyrosine kinase; RDH12, retinol dehydrogenase 12.
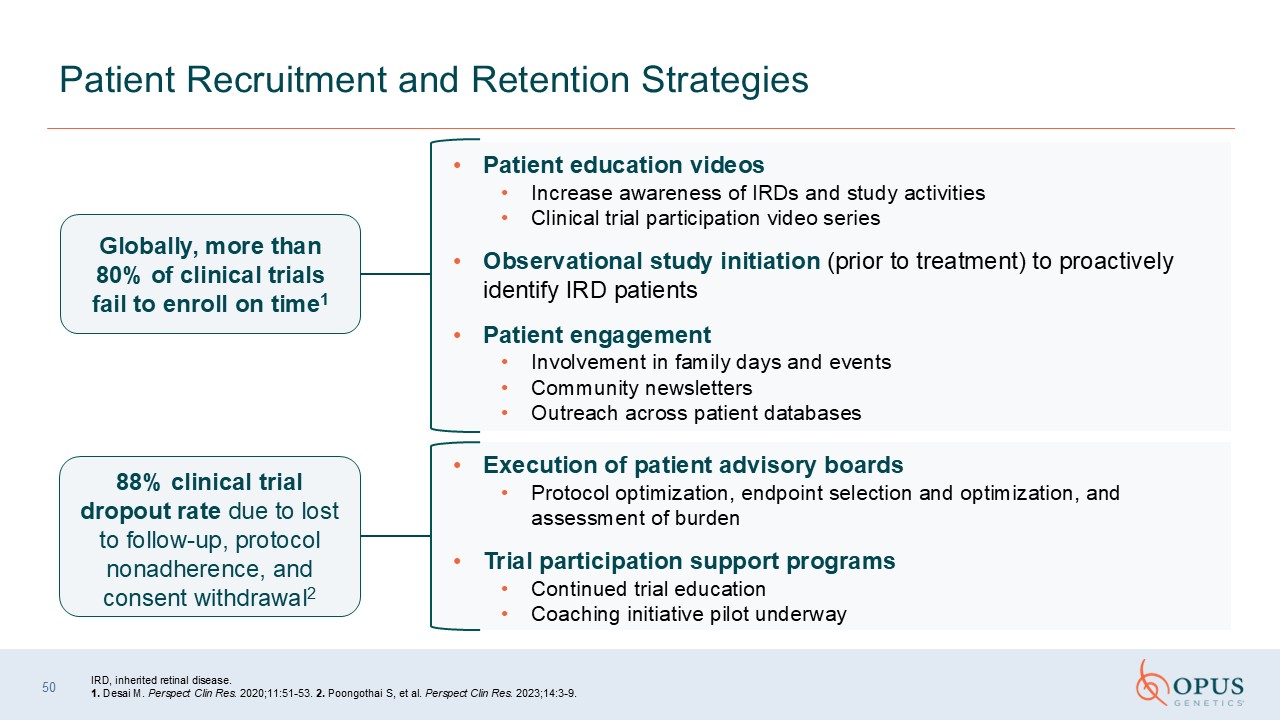
Patient Recruitment and Retention Strategies Patient education
videos Increase awareness of IRDs and study activities Clinical trial participation video series Observational study initiation (prior to treatment) to proactively identify IRD patients Patient engagement Involvement in family days and
events Community newsletters Outreach across patient databases 88% clinical trial dropout rate due to lost to follow‑up, protocol nonadherence, and consent withdrawal2 Globally, more than 80% of clinical trials fail to enroll on
time1 IRD, inherited retinal disease. 1. Desai M. Perspect Clin Res. 2020;11:51-53. 2. Poongothai S, et al. Perspect Clin Res. 2023;14:3-9. Execution of patient advisory boards Protocol optimization, endpoint selection and optimization,
and assessment of burden Trial participation support programs Continued trial education Coaching initiative pilot underway 50

51 Patient Commitment to Expanding Awareness of Disease & Treatment

Q&A Moderated by Ben Yerxa President Opus Genetics

OPGx-LCA5 Update Bart Leroy, MD, PhD Ghent University Hospital Ghent,
Belgium
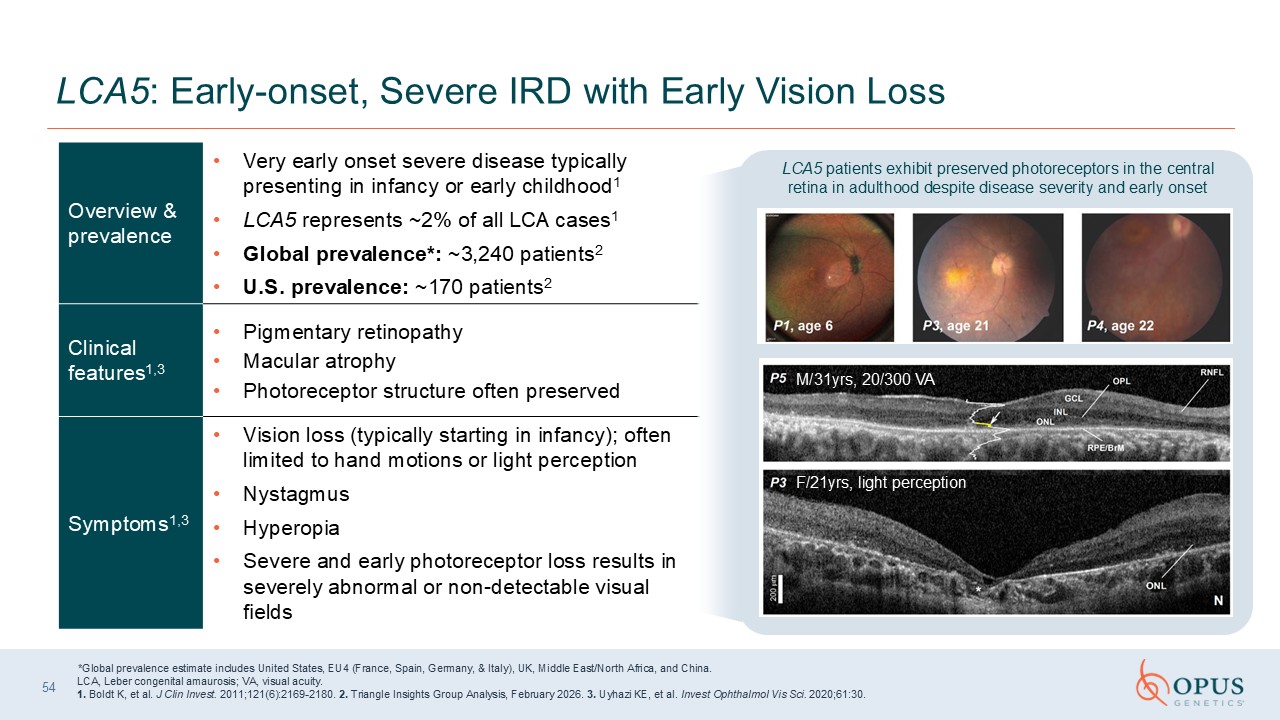
LCA5: Early-onset, Severe IRD with Early Vision Loss Overview &
prevalence Very early onset severe disease typically presenting in infancy or early childhood1 LCA5 represents ~2% of all LCA cases1 Global prevalence*: ~3,240 patients2 U.S. prevalence: ~170 patients2 Clinical features1,3 Pigmentary
retinopathy Macular atrophy Photoreceptor structure often preserved Symptoms1,3 Vision loss (typically starting in infancy); often limited to hand motions or light perception Nystagmus Hyperopia Severe and early photoreceptor loss
results in severely abnormal or non-detectable visual fields *Global prevalence estimate includes United States, EU4 (France, Spain, Germany, & Italy), UK, Middle East/North Africa, and China. LCA, Leber congenital amaurosis; VA, visual
acuity. 1. Boldt K, et al. J Clin Invest. 2011;121(6):2169-2180. 2. Triangle Insights Group Analysis, February 2026. 3. Uyhazi KE, et al. Invest Ophthalmol Vis Sci. 2020;61:30. M/31yrs, 20/300 VA F/21yrs, light perception LCA5 patients
exhibit preserved photoreceptors in the central retina in adulthood despite disease severity and early onset 54

OPGx-LCA5 Gene Therapy is Designed to Restore a Key Protein of the Visual Cycle
OPGx-LCA5 Gene Therapy Vector AAV8 Delivery Single subretinal injection 55 Lebercilin is a ciliary protein critical for the function of photoreceptor inner and outer segments1 In LCA5 patients, photoreceptor function is severely
impaired due to a lack of functioning lebercilin1 However, photoreceptors can survive through the third decade of life, suggestive of a broad window for therapeutic intervention2 OPGx-LCA5 is designed to address mutations in the LCA5 gene,
which encodes for the lebercilin protein Clinically derisked AAV8 vector delivers a functional LCA5 gene directly to photoreceptor cells, using same promoter technology as LUXTURNA® AAV8, adeno-associated virus serotype 8; LCA5, Leber
congenital amaurosis 5. 1. Uyhazi KE, et al. Invest Ophthalmol Vis Sci. 2020;61:30. 2. Song JY, et al. Mol Ther. 2018;26:1581-1593.
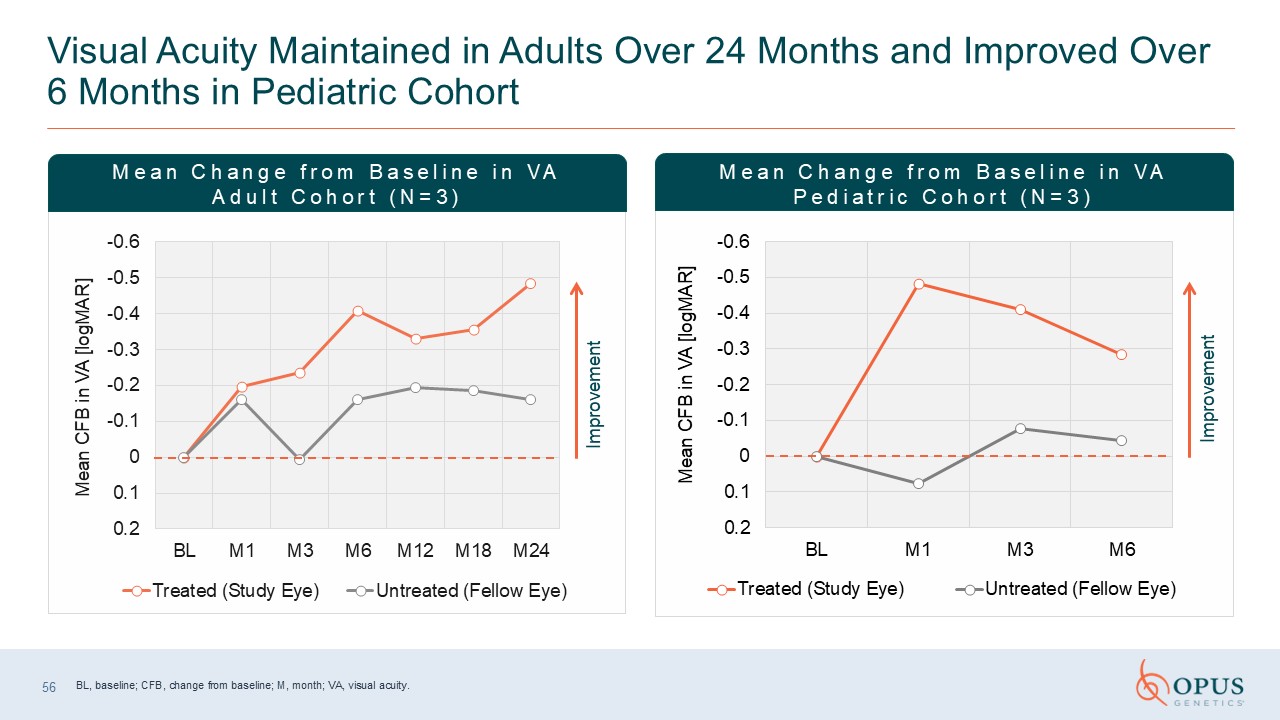
Visual Acuity Maintained in Adults Over 24 Months and Improved Over 6 Months in
Pediatric Cohort Improvement Improvement Mean Change from Baseline in VA Adult Cohort (N=3) Mean Change from Baseline in VA Pediatric Cohort (N=3) BL, baseline; CFB, change from baseline; M, month; VA, visual acuity. 56
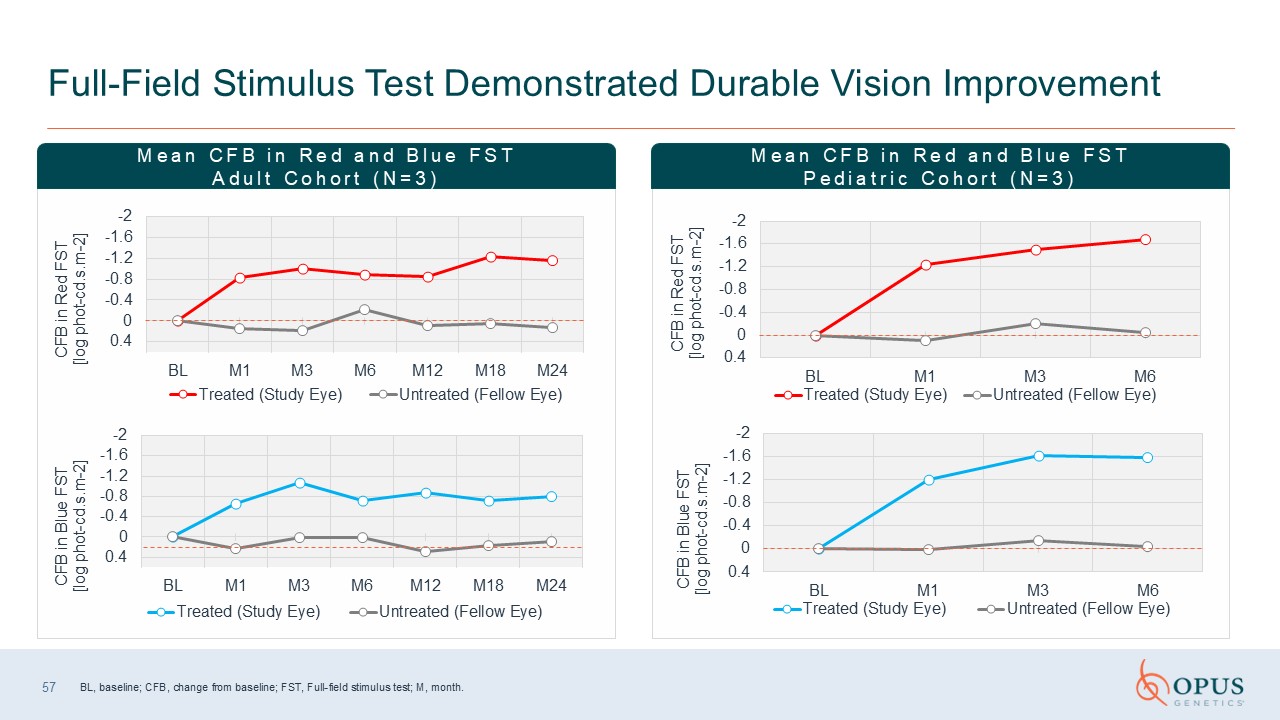
Full-Field Stimulus Test Demonstrated Durable Vision Improvement BL, baseline;
CFB, change from baseline; FST, Full-field stimulus test; M, month. Mean CFB in Red and Blue FST Adult Cohort (N=3) Mean CFB in Red and Blue FST Pediatric Cohort (N=3) 57
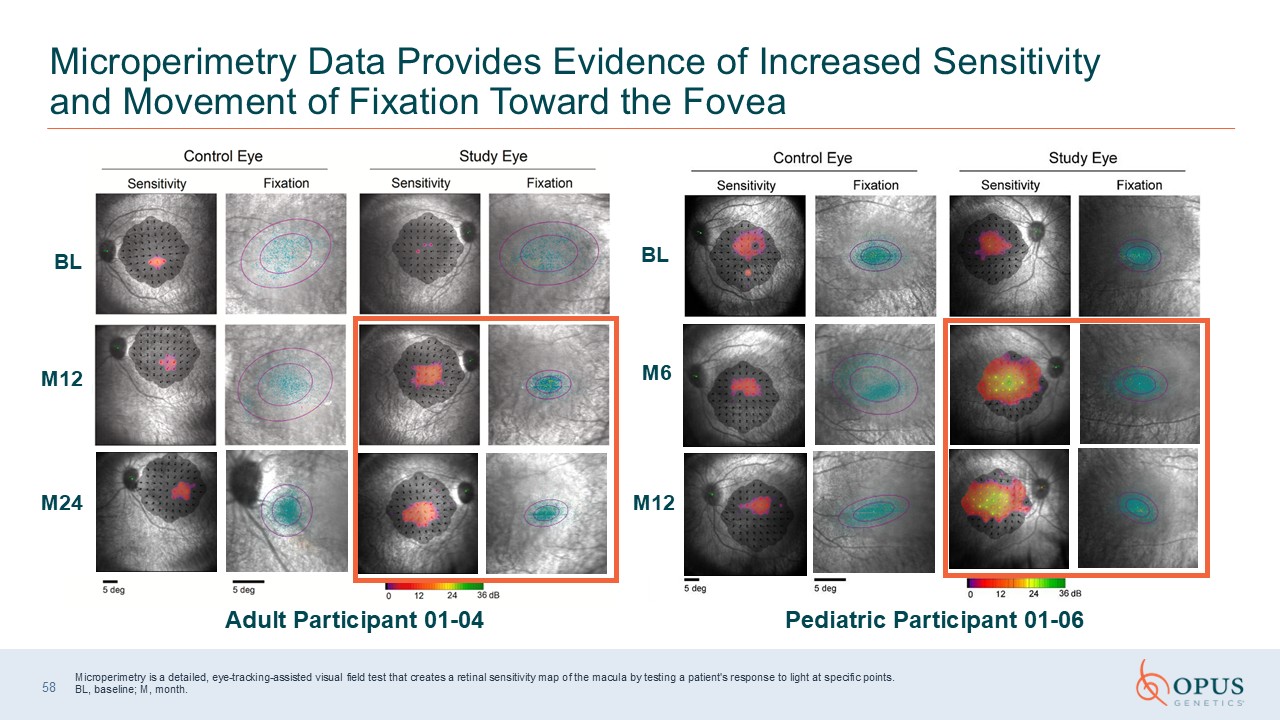
Microperimetry Data Provides Evidence of Increased Sensitivity and Movement of
Fixation Toward the Fovea Adult Participant 01-04 Pediatric Participant 01-06 Microperimetry is a detailed, eye-tracking-assisted visual field test that creates a retinal sensitivity map of the macula by testing a patient's response to
light at specific points.BL, baseline; M, month. 58 BL M12 M24 BL M6 M12

Powerful Participant-Reported Outcomes After OPGx-LCA5 Treatment 59 Adult
Participants 01-01: Reported being able to identify her children within a larger group of children 1 month after surgery which she previously could not have done. 01-03: Had no formative vision prior to treatment. Reported being able to see
his newborn niece for the first time and watch his sister get married. Described his newfound independence, including his ability to pour a glass of wine and drink it unassisted for the first time in his life. Has been able to travel
independently and, as a result, he acquired a new job, which requires a greater level of independence than he previously had. 01-04*: Reported that since treatment, she no longer requires a cane and can now navigate urban environments
independently. Pediatric Participants 01-05: Reported being able to walk and cook without the assistance of others, and how treatment has helped her in her writing capabilities. 01-06*: Reported a noticeable difference in the visual
brightness between his treated and untreated eyes. Able to watch basketball and can see the players and follow the ball instead of watching the score ticker and listening. 01-07: Mostly nonvisual prior to treatment. Reported visiting a local
zoo, where she was able to visualize an owl for the first time, and also reported having to learn colors and what a window was in a hotel Participant-reported outcomes provided in follow up visits in 2025. *01-04 and 01-06 were able to
complete microperimetry.

BEST1 Update Ash Jayagopal, PhD, MBA Chief Scientific and Development Officer
Opus Genetics
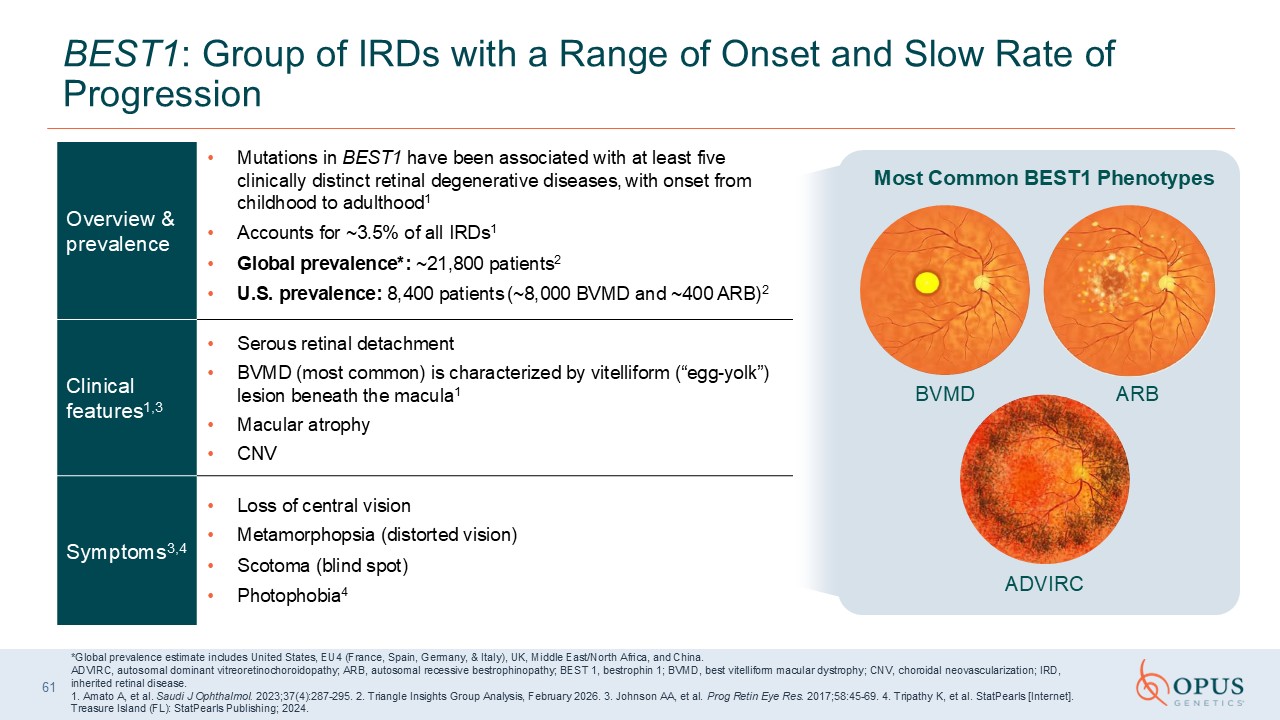
61 BEST1: Group of IRDs with a Range of Onset and Slow Rate of
Progression Overview & prevalence Mutations in BEST1 have been associated with at least five clinically distinct retinal degenerative diseases, with onset from childhood to adulthood1 Accounts for ~3.5% of all IRDs1 Global
prevalence*: ~21,800 patients2 U.S. prevalence: 8,400 patients (~8,000 BVMD and ~400 ARB)2 Clinical features1,3 Serous retinal detachment BVMD (most common) is characterized by vitelliform (“egg-yolk”) lesion beneath the macula1 Macular
atrophy CNV Symptoms3,4 Loss of central vision Metamorphopsia (distorted vision) Scotoma (blind spot) Photophobia4 *Global prevalence estimate includes United States, EU4 (France, Spain, Germany, & Italy), UK, Middle East/North
Africa, and China. ADVIRC, autosomal dominant vitreoretinochoroidopathy; ARB, autosomal recessive bestrophinopathy; BEST 1, bestrophin 1; BVMD, best vitelliform macular dystrophy; CNV, choroidal neovascularization; IRD, inherited retinal
disease. 1. Amato A, et al. Saudi J Ophthalmol. 2023;37(4):287-295. 2. Triangle Insights Group Analysis, February 2026. 3. Johnson AA, et al. Prog Retin Eye Res. 2017;58:45-69. 4. Tripathy K, et al. StatPearls [Internet]. Treasure Island
(FL): StatPearls Publishing; 2024. BVMD ARB ADVIRC Most Common BEST1 Phenotypes
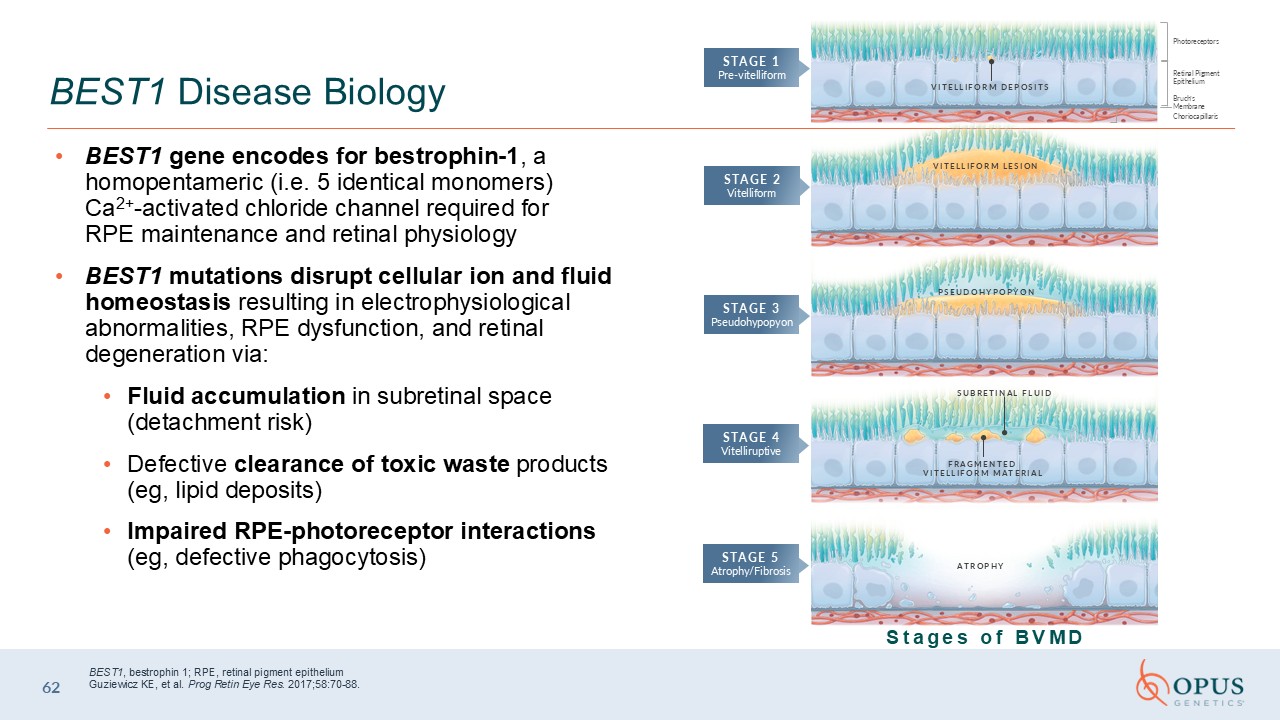
BEST1 gene encodes for bestrophin-1, a homopentameric (i.e. 5 identical
monomers) Ca2+-activated chloride channel required for RPE maintenance and retinal physiology BEST1 mutations disrupt cellular ion and fluid homeostasis resulting in electrophysiological abnormalities, RPE dysfunction, and retinal
degeneration via: Fluid accumulation in subretinal space (detachment risk) Defective clearance of toxic waste products (eg, lipid deposits) Impaired RPE-photoreceptor interactions (eg, defective phagocytosis) 62 BEST1 Disease
Biology BEST1, bestrophin 1; RPE, retinal pigment epitheliumGuziewicz KE, et al. Prog Retin Eye Res. 2017;58:70-88. ATROPHY FRAGMENTED VITELLIFORM MATERIAL SUBRETINAL FLUID PSEUDOHYPOPYON Photoreceptors Retinal Pigment
Epithelium Bruch’s Membrane Choriocapillaris VITELLIFORM LESION VITELLIFORM DEPOSITS STAGE 1 Pre-vitelliform STAGE 2 Vitelliform STAGE 3 Pseudohypopyon STAGE 4 Vitelliruptive STAGE 5 Atrophy/Fibrosis Stages of BVMD
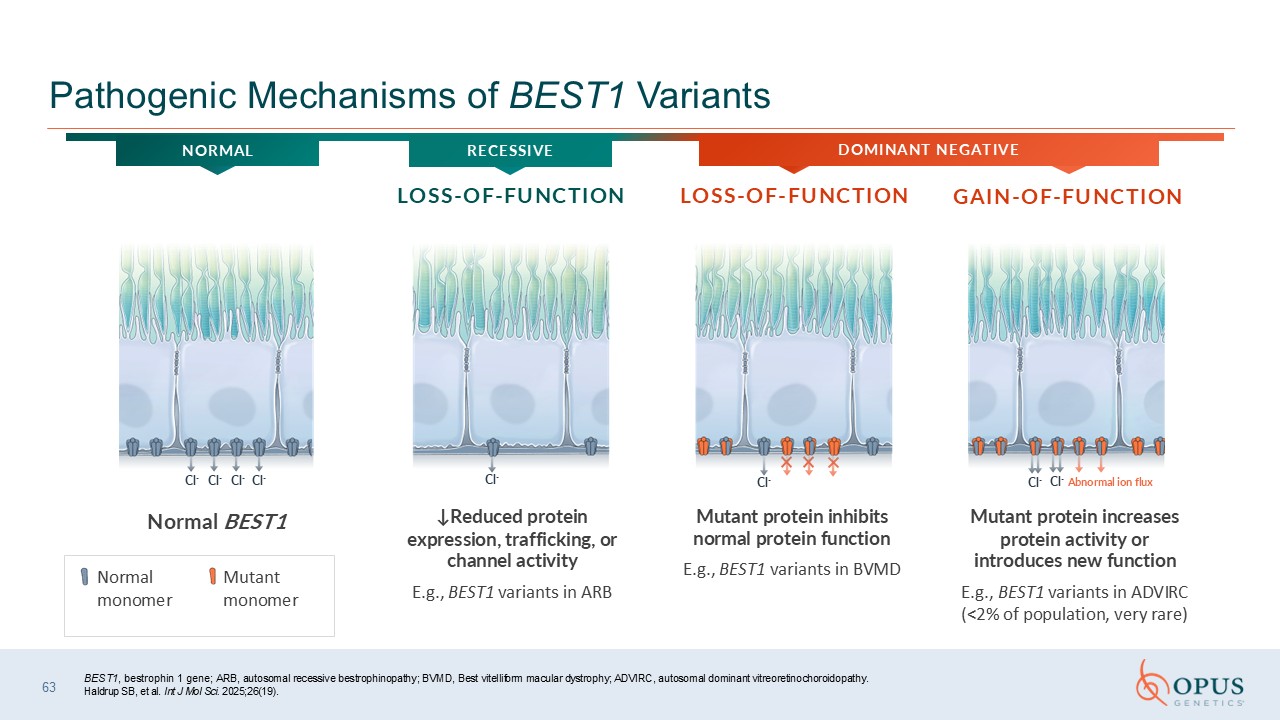
BEST1, bestrophin 1 gene; ARB, autosomal recessive bestrophinopathy; BVMD, Best
vitelliform macular dystrophy; ADVIRC, autosomal dominant vitreoretinochoroidopathy. Haldrup SB, et al. Int J Mol Sci. 2025;26(19). 63 Pathogenic Mechanisms of BEST1 Variants ↓Reduced protein expression, trafficking, or channel
activity E.g., BEST1 variants in ARB Mutant protein inhibits normal protein function E.g., BEST1 variants in BVMD Mutant protein increases protein activity or introduces new function E.g., BEST1 variants in ADVIRC (<2% of population,
very rare) Cl- Cl- Cl- Cl- Cl- Cl- Abnormal ion flux Cl- Cl- GAIN-OF-FUNCTION LOSS-OF-FUNCTION LOSS-OF-FUNCTION DOMINANT NEGATIVE NORMAL Normal monomer Mutant monomer Normal BEST1 RECESSIVE
Cl- Cl- Cl- Cl- Cl- Cl- Cl- Cl- Abnormal ion
flux Cl- Cl- Cl- Cl- Cl- Cl- Cl- BEST1, bestrophin 1 gene; ARB, autosomal recessive bestrophinopathy; BVMD, Best vitelliform macular dystrophy; ADVIRC, autosomal dominant vitreoretinochoroidopathy. Haldrup SB, et al. Int J Mol Sci.
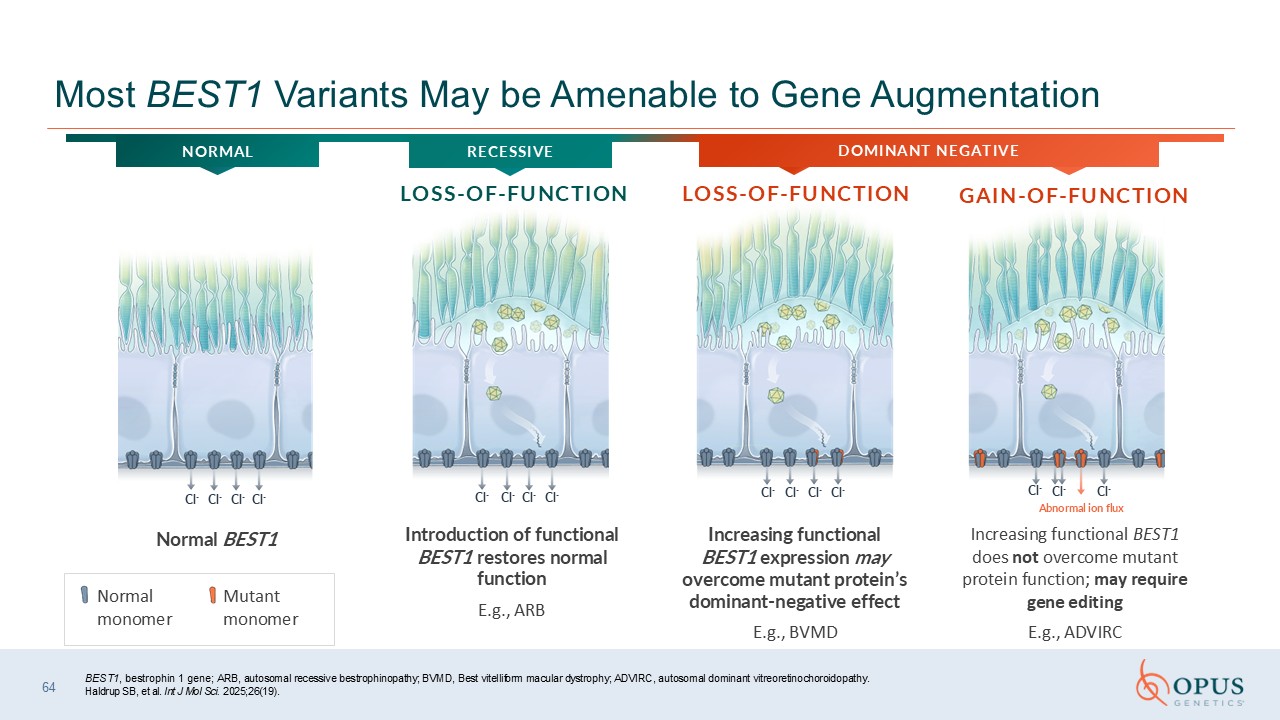
2025;26(19). 64 Most BEST1 Variants May be Amenable to Gene Augmentation Normal monomer Mutant monomer GAIN-OF-FUNCTION LOSS-OF-FUNCTION LOSS-OF-FUNCTION RECESSIVE DOMINANT NEGATIVE NORMAL Introduction of functional BEST1 restores
normal function E.g., ARB Increasing functional BEST1 expression may overcome mutant protein’s dominant-negative effect E.g., BVMD Increasing functional BEST1 does not overcome mutant protein function; may require gene editing E.g.,
ADVIRC Normal BEST1
*Treatment-response assessment conducted on applicable mutations. BEST1,
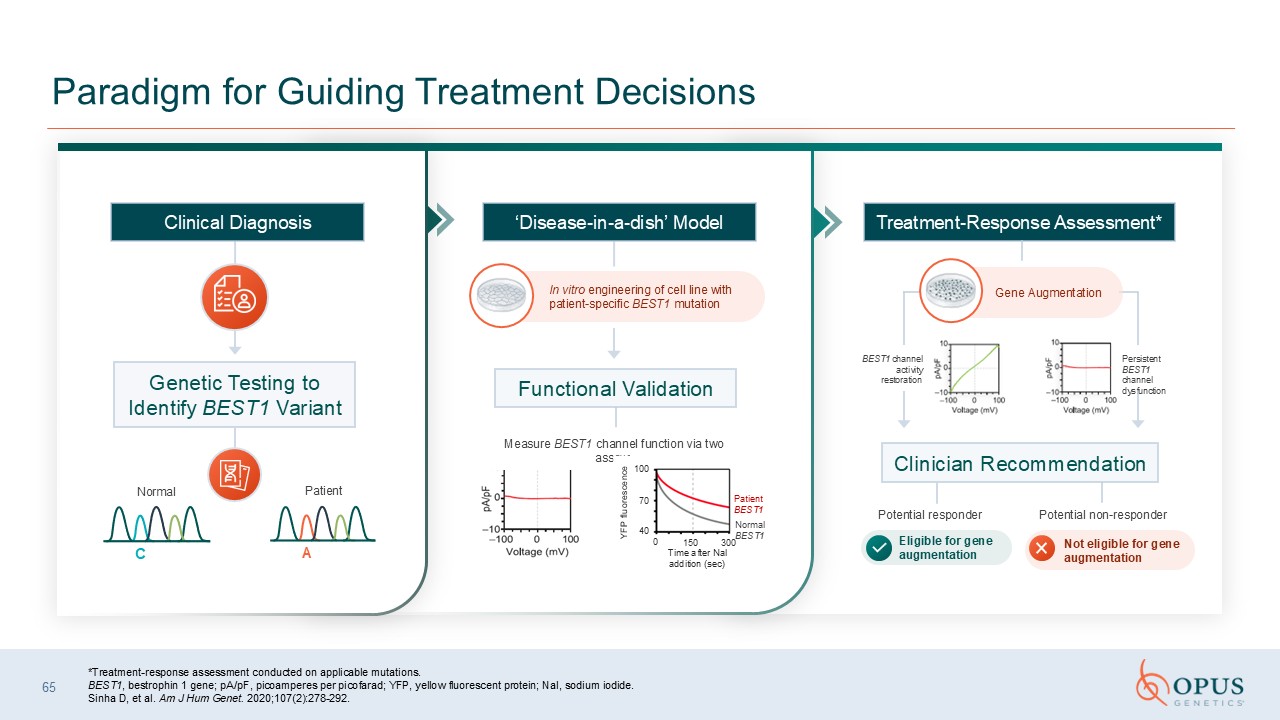
bestrophin 1 gene; pA/pF, picoamperes per picofarad; YFP, yellow fluorescent protein; NaI, sodium iodide. Sinha D, et al. Am J Hum Genet. 2020;107(2):278-292. 65 Paradigm for Guiding Treatment Decisions In vitro engineering of cell line
with patient-specific BEST1 mutation Measure BEST1 channel function via two assays Potential responder Potential non-responder Eligible for gene augmentation Clinical Diagnosis Genetic Testing to Identify BEST1 Variant Functional
Validation Treatment-Response Assessment* Clinician Recommendation ‘Disease-in-a-dish’ Model BEST1 channel activity restoration Gene Augmentation Persistent BEST1 channel dysfunction Not eligible for gene augmentation YFP fluorescence
100 70 40 0 150 300 Time after Nal addition (sec) Normal BEST1 Patient BEST1 Normal Patient C A

Designed to restore retinal ion homeostasis in bestrophinopathies, ameliorating
retinal structural and functional deficits BEST1 is targeted using the AAV2 capsid employed in LUXTURNA® and an RPE-specific promoter Most bestrophinopathies exhibit a slow rate of decline and central photoreceptors usually remain viable
for decades, providing a wide therapeutic window OPGx-BEST1 Gene Therapy is Designed to Target RPE and Restore Bestrophin Function OPGx-BEST1 Gene Therapy Vector AAV2 Delivery Single subretinal injection 66 AAV8, adeno-associated
virus serotype 8; RK1, rhodopsin Kinase 1; RPE, retinal pigment epithelium.

OPGx-BEST1 Clinical Update George Magrath, MD Chief Executive Officer Opus
Genetics
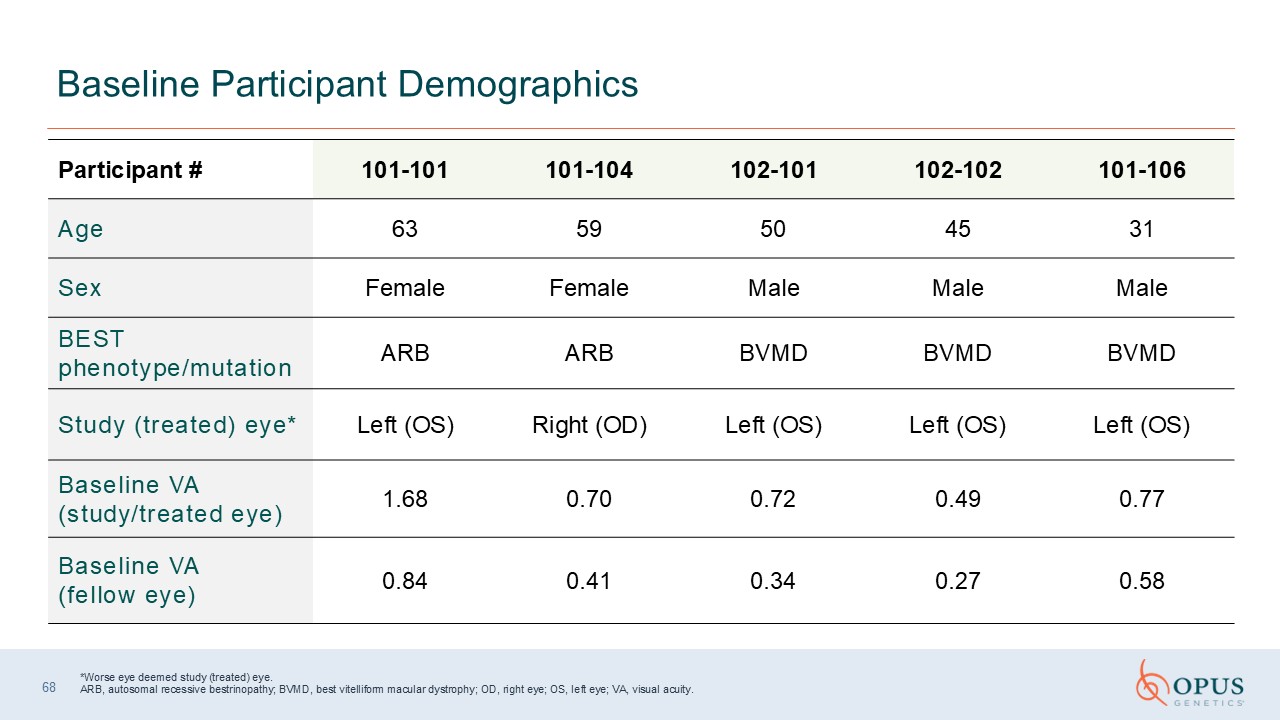
*Worse eye deemed study (treated) eye.ARB, autosomal recessive bestrinopathy;
BVMD, best vitelliform macular dystrophy; OD, right eye; OS, left eye; VA, visual acuity. Baseline Participant Demographics Participant
# 101-101 101-104 102-101 102-102 101-106 Age 63 59 50 45 31 Sex Female Female Male Male Male BEST phenotype/mutation ARB ARB BVMD BVMD BVMD Study (treated) eye* Left (OS) Right (OD) Left (OS) Left (OS) Left
(OS) Baseline VA (study/treated eye) 1.68 0.70 0.72 0.49 0.77 Baseline VA (fellow eye) 0.84 0.41 0.34 0.27 0.58 68
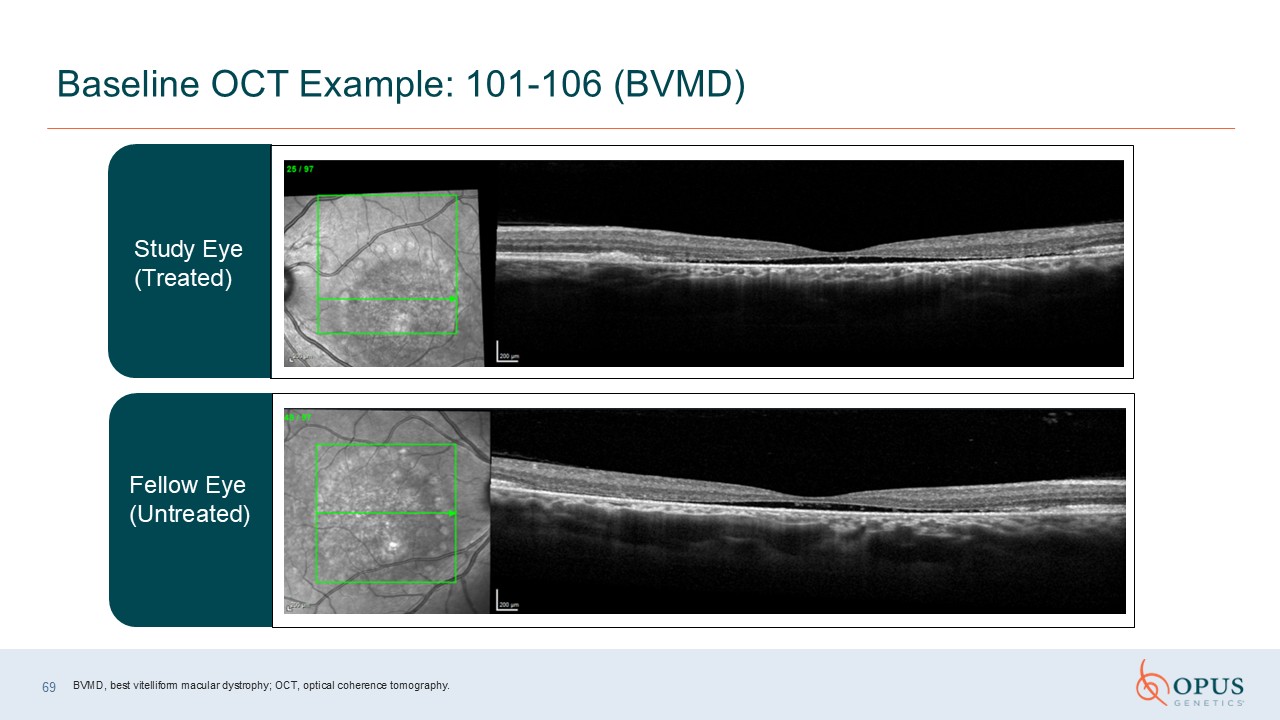
69 Baseline OCT Example: 101-106 (BVMD) Study Eye(Treated) Fellow Eye
(Untreated) BVMD, best vitelliform macular dystrophy; OCT, optical coherence tomography.
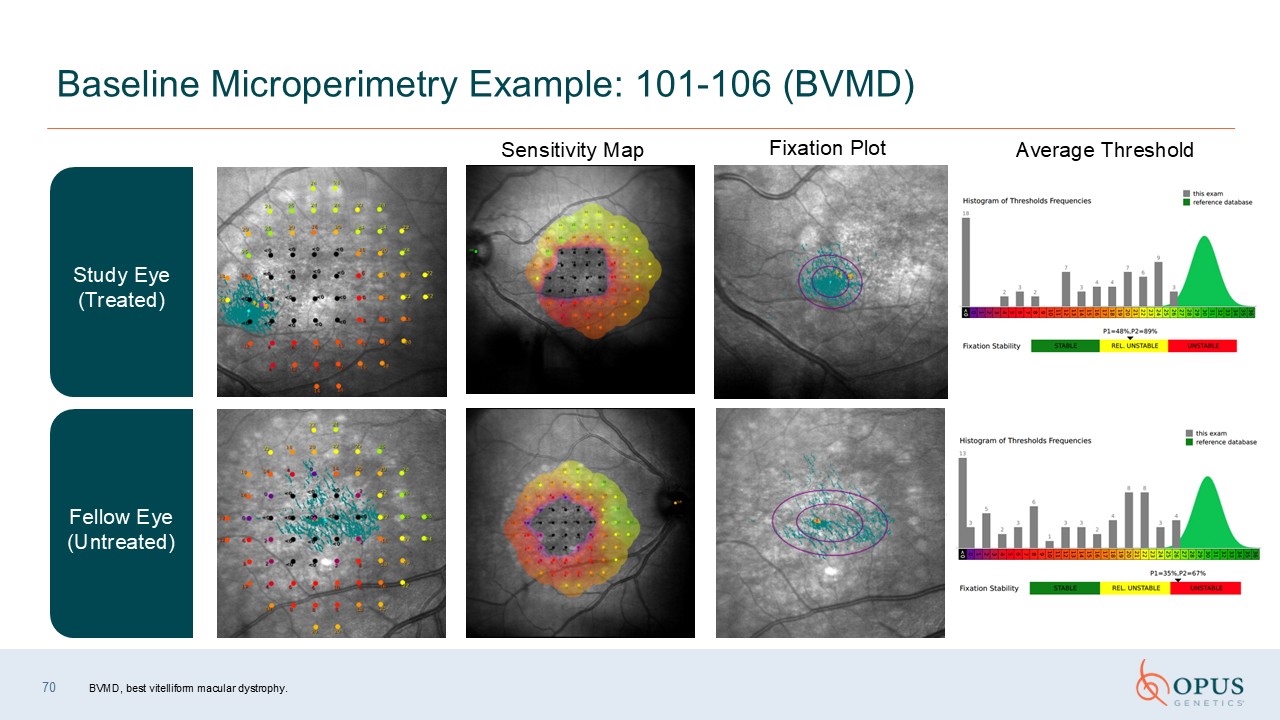
70 Baseline Microperimetry Example: 101-106 (BVMD) Fellow Eye
(Untreated) Study Eye (Treated) Sensitivity Map Fixation Plot Average Threshold BVMD, best vitelliform macular dystrophy.

IRD Patient Journey & Disease Prevalence Joe Schachle, MBA Chief Operating
Officer Opus Genetics
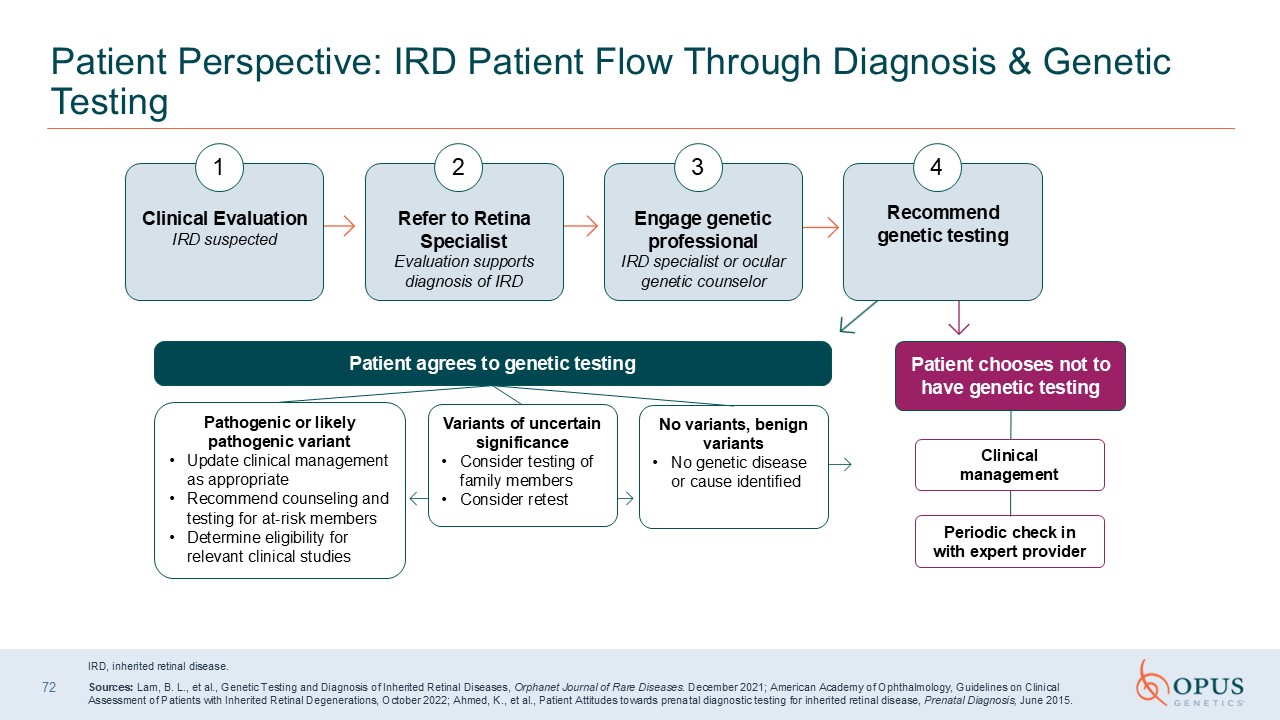
72 Patient Perspective: IRD Patient Flow Through Diagnosis & Genetic
Testing IRD, inherited retinal disease. Sources: Lam, B. L., et al., Genetic Testing and Diagnosis of Inherited Retinal Diseases, Orphanet Journal of Rare Diseases. December 2021; American Academy of Ophthalmology, Guidelines on Clinical
Assessment of Patients with Inherited Retinal Degenerations, October 2022; Ahmed, K., et al., Patient Attitudes towards prenatal diagnostic testing for inherited retinal disease, Prenatal Diagnosis, June 2015. Clinical Evaluation IRD
suspected 1 Refer to Retina SpecialistEvaluation supports diagnosis of IRD 2 Engage genetic professionalIRD specialist or ocular genetic counselor 3 Recommend genetic testing 4 Patient agrees to genetic testing Pathogenic or likely
pathogenic variant Update clinical management as appropriate Recommend counseling and testing for at-risk members Determine eligibility for relevant clinical studies Variants of uncertain significance Consider testing of family
members Consider retest No variants, benign variants No genetic disease or cause identified Patient chooses not to have genetic testing Clinical management Periodic check in with expert provider
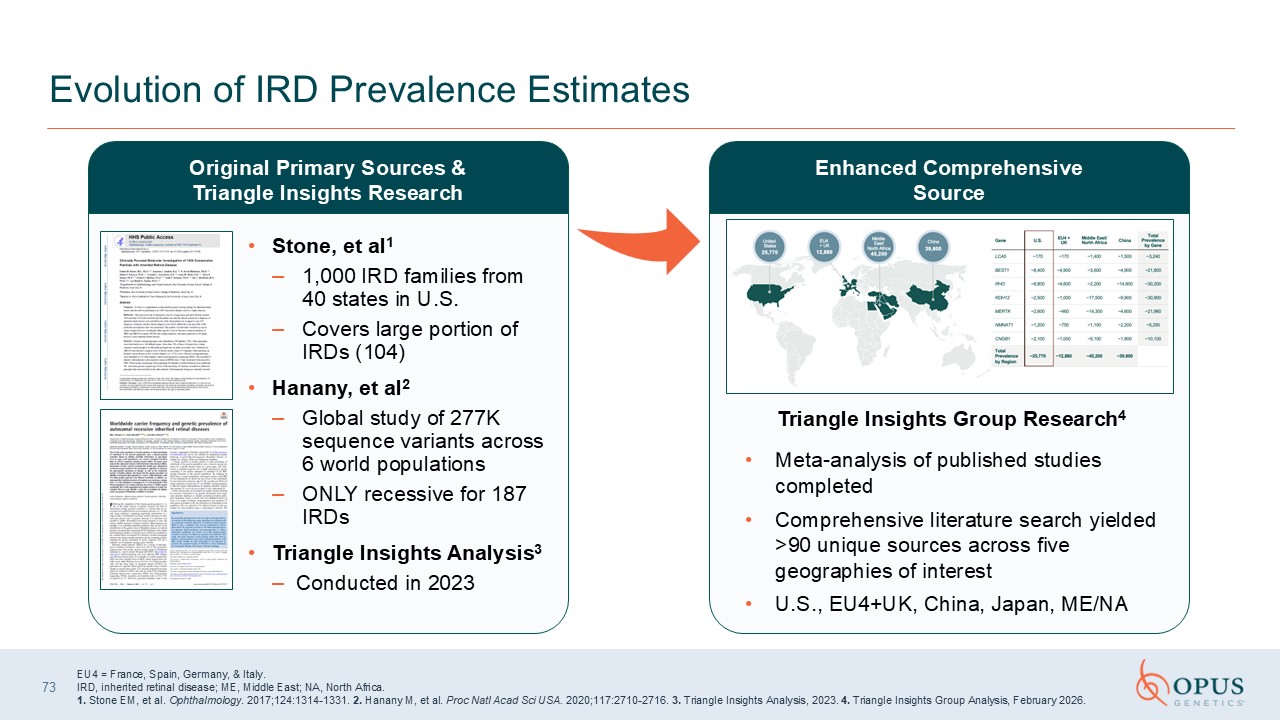
Stone, et al1 1,000 IRD families from 40 states in U.S. Covers large portion
of IRDs (104) Hanany, et al2 Global study of 277K sequence variants across 6 world populations ONLY recessive for 187 IRDs Triangle Insights Analysis3 Conducted in 2023 73 Evolution of IRD Prevalence Estimates Triangle Insights Group
Research4 Meta-analysis of published studies completed Comprehensive literature search yielded >90 unique sources across five geographies of interest U.S., EU4+UK, China, Japan, ME/NA Original Primary Sources & Triangle Insights
Research Enhanced Comprehensive Source EU4 = France, Spain, Germany, & Italy. IRD, inherited retinal disease; ME, Middle East; NA, North Africa. 1. Stone EM, et al. Ophthalmology. 2017;124:1314-1331. 2. Hanany M, et al. Proc Natl
Acad Sci USA. 2020;117:2710-2716. 3. Triangle Insights Analysis, 2023. 4. Triangle Insights Group Analysis, February 2026.
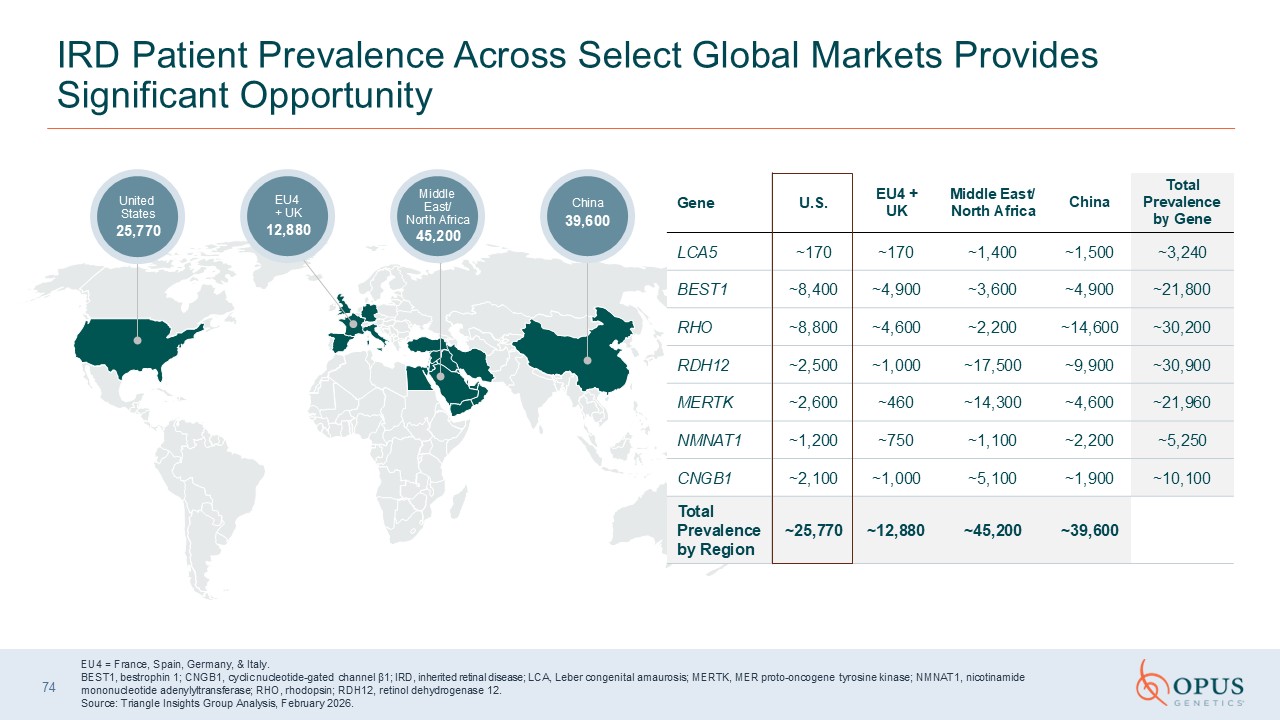
IRD Patient Prevalence Across Select Global Markets Provides Significant
Opportunity China 39,600 Middle East/North Africa 45,200 EU4 + UK 12,880 United States 25,770 EU4 = France, Spain, Germany, & Italy. BEST1, bestrophin 1; CNGB1, cyclic nucleotide-gated channel β1; IRD, inherited retinal disease;
LCA, Leber congenital amaurosis; MERTK, MER proto-oncogene tyrosine kinase; NMNAT1, nicotinamide mononucleotide adenylyltransferase; RHO, rhodopsin; RDH12, retinol dehydrogenase 12. Source: Triangle Insights Group Analysis, February
2026. Gene U.S. EU4 + UK Middle East/North Africa China Total Prevalenceby
Gene LCA5 ~170 ~170 ~1,400 ~1,500 ~3,240 BEST1 ~8,400 ~4,900 ~3,600 ~4,900 ~21,800 RHO ~8,800 ~4,600 ~2,200 ~14,600 ~30,200 RDH12 ~2,500 ~1,000 ~17,500 ~9,900 ~30,900 MERTK ~2,600 ~460 ~14,300 ~4,600 ~21,960 NMNAT1 ~1,200 ~750 ~1,100 ~2,200 ~5,250 CNGB1 ~2,100 ~1,000 ~5,100 ~1,900 ~10,100 Total
Prevalence by Region ~25,770 ~12,880 ~45,200 ~39,600 74

IRD Prevalence May Be Higher Than Current Estimates BEST1 Prevalence Analysis
based on published studies with diagnostic testing to confirm diagnosis* Estimated 8,400 Patients in the U.S. with BEST1 Patients may not undergo genetic testing when clinicians diagnose BEST1 disease based on the presence of a
characteristic vitelliform (“egg-yolk”) lesion beneath the macula Misdiagnosis may contribute to underestimation of BEST1 prevalence “Quite a few of my BEST patients have been seen by other physicians in my practice and did not get the
diagnosis of BEST disease. I think there are a lot of patients who are not being diagnosed correctly… I would say about 50%.” - Retinal Specialist BEST1, bestrophin 1; IRD, inherited retinal disease. Source: Triangle Insights Group
Analysis, February 2026. BEST1 Disease is Likely Underreported 75

Patient Recruitment &Retention Panel Moderator: Ben Yerxa Panelists: Jean
Bennett, MD, PhD Todd Durham, PhD Bart Leroy, MD, PhD

Q&A

Summary Takeaways George Magrath, MD, MBA, MS Chief Executive Officer
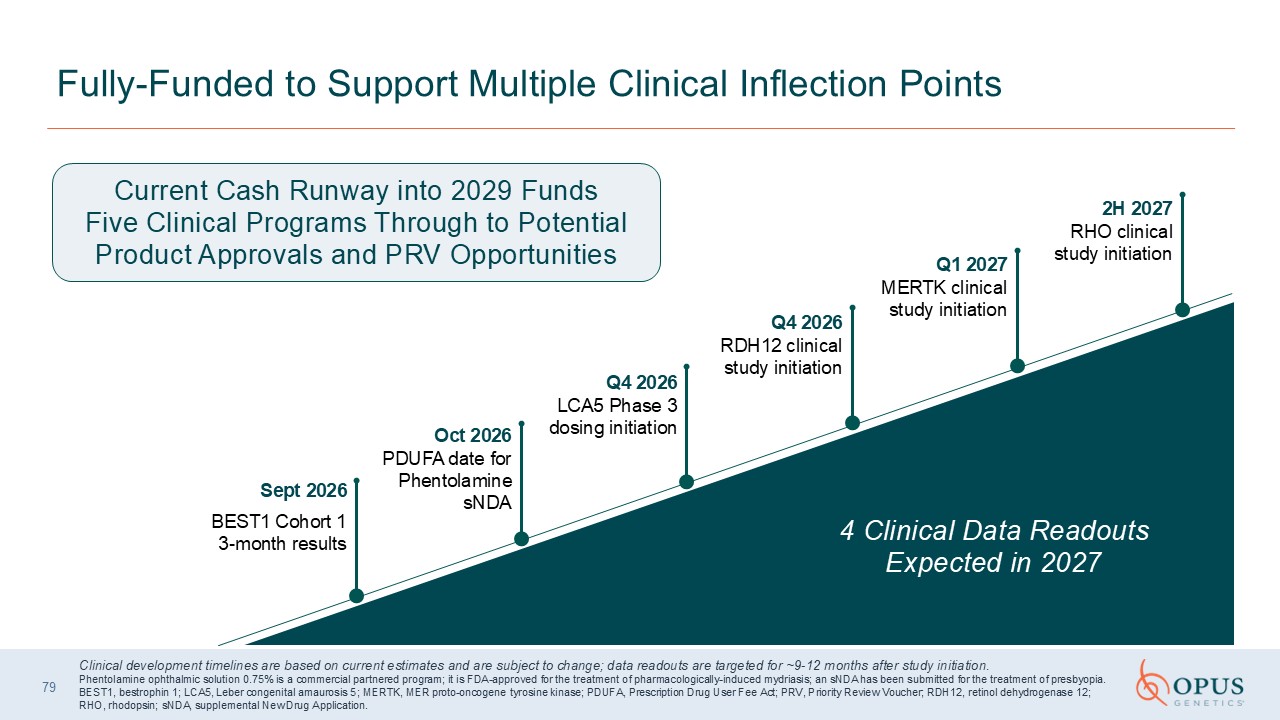
79 Fully-Funded to Support Multiple Clinical Inflection Points 2H 2027 RHO
clinical study initiation Q1 2027 MERTK clinical study initiation Q4 2026RDH12 clinical study initiation Q4 2026LCA5 Phase 3 dosing initiation Oct 2026PDUFA date for Phentolamine sNDA Sept 2026 BEST1 Cohort 1 3-month results Current
Cash Runway into 2029 Funds Five Clinical Programs Through to Potential Product Approvals and PRV Opportunities 4 Clinical Data Readouts Expected in 2027 Clinical development timelines are based on current estimates and are subject to
change; data readouts are targeted for ~9-12 months after study initiation. Phentolamine ophthalmic solution 0.75% is a commercial partnered program; it is FDA-approved for the treatment of pharmacologically-induced mydriasis; an sNDA has
been submitted for the treatment of presbyopia. BEST1, bestrophin 1; LCA5, Leber congenital amaurosis 5; MERTK, MER proto-oncogene tyrosine kinase; PDUFA, Prescription Drug User Fee Act; PRV, Priority Review Voucher; RDH12, retinol
dehydrogenase 12; RHO, rhodopsin; sNDA, supplemental New Drug Application.

80 Thank you