10-K: Annual report pursuant to Section 13 and 15(d)
Published on
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☑ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE
SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2017
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d)
OF
THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File No.:001-34079
Rexahn Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware
|
11-3516358
|
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification Number)
|
15245 Shady Grove Road, Suite 455
Rockville, MD 20850
(Address of principal executive offices, including zip code)
Telephone: (240) 268-5300
(Registrant’s telephone number, including area code)
|
Title of Each Class
|
Name of Each Exchange On Which Registered
|
|
|
Common Stock, $0.0001 par value per share
|
NYSE American
|
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined by Rule-405 of the Securities Act. Yes ☐ No ☑
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
Yes ☐ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein; and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “accelerated filer,” “large accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large Accelerated Filer
|
☐
|
Accelerated Filer
|
☑
|
|
Non-Accelerated Filer
|
☐
|
Smaller reporting company
|
☐
|
|
(Do not check if a smaller reporting company)
|
Emerging growth company
|
☐
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act) Yes ☐ No ☑
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter: As of June 30, 2017, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was $79,303,524 based on the closing price reported on NYSE American.
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date:
|
Class
|
Outstanding as of March 9, 2018
|
|
|
Common Stock, $0.0001 par value per share
|
31,744,439 shares
|
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant’s Definitive Proxy Statement for its 2018 Annual Meeting of Stockholders, which is expected to be filed with the U.S. Securities and Exchange Commission within 120 days after the end of the registrant’s fiscal year ended December 31, 2017, are incorporated by reference into Part III of this Annual Report on Form 10-K.
Cautionary Statement Regarding Forward-Looking Statements.
This Annual Report on Form 10‑K contains statements (including certain projections and business trends) accompanied by such phrases as “believe,” “estimate,” “expect,” “anticipate,” “will,” “intend” and other similar expressions, that are “forward-looking statements” as defined in the Private Securities Litigation Reform Act of 1995. We caution that forward-looking statements are based largely on our expectations and are subject to a number of known and unknown risks and uncertainties that are subject to change based on factors that are, in many instances, beyond our control. Actual results, performance or achievements may differ materially from those contemplated, expressed or implied by the forward-looking statements.
Although we believe that the expectations reflected in our forward-looking statements are reasonable as of the date we make them, actual results could differ materially from those currently anticipated due to a number of factors, including risks relating to:
| · |
our understandings and beliefs regarding the role of certain biological mechanisms and processes in cancer;
|
| · |
our drug candidates being in early stages of development, including in pre-clinical development;
|
| · |
our ability to initially develop drug candidates for orphan indications to reduce the time-to-market and take advantage of certain incentives provided by the U.S. Food and Drug Administration;
|
| · |
our ability to transition from our initial focus on developing drug candidates for orphan indications to candidates for more highly prevalent indications;
|
| · |
our ability to successfully and timely complete clinical trials for our drug candidates in clinical development;
|
| · |
uncertainties related to the timing, results and analyses related to our drug candidates in pre-clinical development;
|
| · |
our ability to obtain the necessary U.S. and international regulatory approvals for our drug candidates;
|
| · |
our reliance on third-party contract research organizations and other investigators and collaborators for certain research and development services;
|
| · |
our ability to maintain or engage third-party manufacturers to manufacture, supply, store and distribute supplies of our drug candidates for our clinical trials;
|
| · |
our ability to form strategic alliances and partnerships with pharmaceutical companies and other partners for sales and marketing of certain of our product candidates;
|
| · |
demand for and market acceptance of our drug candidates;
|
| · |
the scope and validity of our intellectual property protection for our drug candidates and our ability to develop our candidates without infringing the intellectual property rights of others;
|
| · |
our lack of profitability and the need for additional capital to operate our business; and
|
| · |
other risks and uncertainties, including those set forth herein under the caption “Risk Factors” and those detailed from time to time in our filings with the Securities and Exchange Commission.
|
These forward-looking statements are made only as of the date hereof, and we undertake no obligation to update or revise the forward-looking statements, whether as a result of new information, future events or otherwise.
REXAHN PHARMACEUTICALS, INC.
|
Page
|
|||
|
PART I
|
1
|
||
|
Item 1
|
1
|
||
|
Item 1A
|
24
|
||
|
Item 1B
|
43
|
||
|
Item 2
|
43
|
||
|
Item 3
|
43
|
||
|
Item 4
|
43
|
||
|
PART II
|
44
|
||
|
Item 5
|
44
|
||
|
Item 6
|
46
|
||
|
Item 7
|
47
|
||
|
Item 7A
|
59
|
||
|
Item 8
|
59
|
||
|
Item 9
|
60
|
||
|
Item 9A
|
60
|
||
|
Item 9B
|
62
|
||
|
PART III
|
63 | ||
|
Item 10
|
63
|
||
|
Item 11
|
63
|
||
|
Item 12
|
63
|
||
|
Item 13
|
63
|
||
|
Item 14
|
63
|
||
|
Item 15
|
64
|
||
|
Item 16
|
64
|
||
|
69
|
|||
PART I
Unless the context requires otherwise, any references in this Annual Report on Form 10-K to “we,” “us,” “our,” the “Company” or “Rexahn” refers to Rexahn Pharmaceuticals, Inc.
Overview
We are a clinical stage biopharmaceutical company dedicated to the discovery and development of innovative treatments for cancer. Our mission is to improve the lives of cancer patients by developing next-generation cancer therapies that are designed to maximize efficacy while minimizing the toxicity and side effects traditionally associated with cancer treatment. Our pipeline features two oncology product candidates in Phase 2 clinical development and additional compounds in pre-clinical development. Our strategy is to continue building a significant pipeline of innovative oncology product candidates that we intend to commercialize with partners. Our clinical stage drug candidates in active development are RX-3117 and RX-5902 (Supinoxin™).
| · |
RX-3117 is a novel, oral, small molecule nucleoside compound. Once intracellularly activated (phosphorylated) by the enzyme UCK2, it is incorporated into the DNA or RNA of cells and inhibits both DNA and RNA synthesis, which induces apoptotic cell death of tumor cells. Because UCK2 is overexpressed in multiple human tumors, but has a very limited presence in normal tissues, RX-3117 offers the potential for a targeted anti-cancer therapy with an improved efficacy and safety profile, and we believe it has therapeutic potential in a broad range of cancers, including pancreatic, bladder, colon, and lung cancer. In January 2018, we reported final data from a Phase 2a clinical trial of RX-3117 in patients with relapsed or refractory metastatic pancreatic cancer. In this trial, encouraging progression free survival and evidence of tumor shrinkage were observed in patients with metastatic pancreatic cancer that was resistant to gemcitabine and who had failed on multiple prior treatments. RX-3117 is currently the subject of a Phase 2a clinical trial in combination with Abraxane® (paclitaxel protein-bound) in patients newly diagnosed with metastatic pancreatic cancer. In February 2018, updated safety and efficacy data from the ongoing Phase 2a clinical trial of RX-3117 in advanced urothelial (bladder) cancer were reported. In this trial, encouraging progression free survival and evidence of tumor shrinkage were observed in patients with advanced bladder cancer who had failed on multiple prior treatments including immunotherapy and gemcitabine. RX-3117 has received “orphan drug designation” from the U.S. Food and Drug Administration (“FDA”) and from the European Commission (“EC”) for pancreatic cancer. Orphan drug designation in the U.S. provides tax incentives for clinical research and a waiver from user fees under certain circumstances. In addition, an orphan drug generally receives seven years of exclusivity in the U.S. after approval for a designated use, during which time the FDA generally cannot approve another product with the same active moiety for the same indication.
|
| · |
RX-5902 (Supinoxin) is a potential first-in-class small molecule inhibitor of phosphorylated-p68, a protein that we believe plays a key role in cancer cell growth, progression and metastasis through its interaction with beta-catenin. Phosphorylated p68, which is highly expressed in cancer cells, but not in normal cells, results in up-regulation of cancer-related genes and a subsequent proliferation of cancer cells and tumor growth. RX-5902 selectively blocks the interaction of phosphorylated p68 with beta-catenin, thereby decreasing the proliferation or growth of cancer cells in preclinical models. In addition, multiple pre-clinical models suggest that RX-5902 enhances the efficacy of immunotherapy. We have evaluated RX-5902 in a Phase 1 dose escalation study in patients with a diverse range of metastatic, treatment-refractory tumors, including breast, ovarian, colorectal, and neuro-endocrine tumors. In February 2017, we initiated a Phase 2a clinical study of RX-5902 in patients with metastatic triple negative breast cancer (“TNBC”).
|
| · |
RX-0201 (Archexin) is a potential best-in-class, potent inhibitor of the protein kinase Akt-1, which we believe plays a critical role in cancer cell proliferation, survival, angiogenesis, metastasis and drug resistance. RX-0201 is the subject of a research and development collaboration with Zhejiang Haichang Biotechnology Co., Ltd. (“Haichang”) for the development of RX-0201 to conduct certain pre-clinical and clinical activities through completion of a Phase 2a proof-of-concept clinical trial in hepatocellular carcinoma (“HCC”) and pursuant to which the parties will share any downstream licensing fees and royalties paid by third parties in connection with the further development and commercialization of RX-0201 for the treatment of HCC. RX-0201 has received orphan drug designation from the FDA for renal cell carcinoma (“RCC”), glioblastoma, ovarian cancer, stomach cancer and pancreatic cancer. In February 2018, in response to the changing treatment landscape for metastatic RCC over the past two years with the approval of new therapies by the FDA, we announced plans to discontinue the internally funded programs of RX-0201 and ceased enrolling patients in a Phase 2a proof-of-concept clinical trial of RX-0201 in patients with metastatic renal cell carcinoma.
|
In addition to our drug development efforts, our nano-based drug delivery systems, such as those used in the multiple nanoliposomal- and nanopolymer-based anti-cancer drugs that we are currently testing, may increase the availability of a drug at the disease site, minimize adverse reactions, and provide longer duration of action.
Company Background
We trace our history to the March 2001 founding of Rexahn, Corp. Dr. Peter D. Suzdak, our Chief Executive Officer since February 2013, has extensive experience in corporate management and drug development, particularly in the field of oncology.
Our common stock is currently listed on the NYSE American under the trading symbol “RNN.” Our principal corporate office is located at 15245 Shady Grove Road, Suite 455, Rockville, Maryland 20850 in Maryland’s I-270 technology corridor. Our telephone number is (240) 268-5300.
Industry and Disease Markets
Market Overview
Our primary research and development focus is oncology therapeutics. A key component of our strategy is to develop innovative drugs that are potential first-in-class or market-leading compounds for the treatment of cancer. According to the Centers for Disease Control and Prevention, cancer claims the lives of more than half a million Americans each year and is the second leading cause of death among Americans. The World Health Organization estimated in 2018 that there were 14 million new cases of cancer diagnosed worldwide in 2012 and that cancer was responsible for 8.8 million deaths in 2015. A 2018 American Cancer Society report projected that an estimated 1.7 million new cancer cases will be diagnosed in the United States in 2018. The IQVIA Institute for Human Data Science, formerly the IMS Institute for Healthcare Informatics, reported in 2017 that total global spending on oncology medicines, including therapeutic treatments and supportive care, reached $113 billion in 2016.
Current Cancer Treatments
Traditional cancer treatments involve surgery, radiation therapy and chemotherapy. Surgery is widely used to treat cancer but may result in related or significant complications and may be ineffective if metastasis has occurred. Radiation therapy, or radiotherapy, can be highly effective in treating certain types of cancer. In radiation therapy, ionizing radiation deposits energy that injures or destroys cells in the area being treated by damaging their genetic material, making it impossible for these cells to continue to grow. Although radiation damages both cancer cells and normal cells, the normal cells are generally able to repair themselves and function properly. Chemotherapy involves the use of cytotoxic cancer drugs to destroy cancer cells by interfering with various stages of the cell division process. For certain cancers and in certain patients, these drugs have limited efficacy and debilitating adverse side effects. Administration of generally cytotoxic cancer drugs may also result in the development of multi-drug resistance, a condition that results when certain tumor cells that have survived treatment with cytotoxic drugs are no longer susceptible to treatment by those and other drugs. Recent advances in cancer treatment include the use of cancer-targeted cytotoxic agents and immunotherapies to stimulate the body’s own immune system to kill cancer cells. Immunotherapy can significantly improve survival in certain cancers, including melanoma, non-small cell lung cancer, head and neck tumors, lymphoma and renal cell carcinoma. However, immunotherapy approaches have not been effective in all tumor types and there is a risk of over-stimulation of the immune system that can lead to life-threatening autoimmune side-effects, such as colitis, pneumonia, and hepatitis.
Unmet Needs in Cancer
Despite significant advances in cancer research and treatments, many unmet needs still remain including:
| · |
Long-term management of cancers: Surgery, radiation therapy or chemotherapy may not result in long-term remission, although surgery and radiation therapies are considered effective methods for some cancers. There is a need for more effective drugs and adjuvant therapies to treat relapsed and refractory cancers.
|
| · |
Multi-drug resistance: Multi-drug resistance is a major obstacle to effectively treating various cancers with chemotherapy.
|
| · |
Debilitating toxicity by chemotherapy: Chemotherapy as a mainstay of cancer treatment can induce severe adverse reactions and toxicities, adversely affecting quality of life or life itself.
|
Market Opportunity
There are several factors that we believe are favorable for commercializing new cancer drugs that may have the potential to be first-in-class or market leaders, including:
| · |
Expedited Regulatory or Commercialization Pathways. Drugs for life-threatening diseases such as cancer are often candidates for fast track designation, breakthrough therapy designation, priority review and accelerated approval, each of which may lead to approval sooner than would otherwise be the case.
|
| · |
Favorable Environment for Formulary Access and Reimbursement. We believe cancer drugs with proven efficacy would gain rapid market uptake, formulary listing and third-party payor reimbursement. Drugs with orphan designations are generally reimbursed by third-party payors because there are few, if any, alternatives.
|
| · |
Low Marketing Costs. We believe the marketing of new drugs to oncologists can be accomplished with a smaller sales force and lower related costs than a sales force that markets widely to primary care physicians and general practitioners.
|
Our Strategy
Our strategy is to continue building a significant product pipeline of innovative drug candidates that we intend to commercialize alone or with partners. This strategy has several key components.
Develop Innovative Therapeutics with the Potential to be First-in-Class or Market Leaders
We plan to focus our research and development pipeline on potential first-in-class or market-leading compounds for the treatment of cancer. By expanding the breadth and depth of our oncology pipeline, we aim to develop an industry-leading oncology therapeutics franchise. Our pipeline spans several major classes of cancer drugs, including molecular targeted therapies and nano-medicines for targeted delivery of compounds and small molecule cytotoxic compounds. Differentiated target product profiles and proprietary discovery and research technology platforms further support these strategic efforts.
Clinically Develop Drug Candidates as Orphan Drugs in Underserved Specialty Oncology Indications
We intend to initially develop drug candidates for cancers that are orphan indications or for indications where there has been very little innovation and a high unmet need for better treatments. Under the Orphan Drug Act, the FDA may grant orphan drug designation to new drugs developed to treat diseases generally affecting less than 200,000 patients. Benefits associated with orphan drug designation include tax incentives for research and development and an exemption from user fees under certain circumstances. Although the standards for orphan drug approval are not different than for non-orphan products, the path to approval may be faster because clinical trials may be smaller due to the smaller patient population. Further, a drug that is approved for its orphan-designated indication generally receives seven years of orphan drug exclusivity, during which the FDA generally may not approve any other application for a product containing the same active moiety and proposed for the same indication. An approved orphan drug also may qualify for an exemption from the branded prescription drug fee. The European Union (“EU”) also has a version of orphan drug designation, which generally carries with it a ten-year period of market exclusivity. We plan to develop drug candidates, when possible, for cancers that are orphan indications to take advantage of the benefits of orphan drug designation during development and the exclusivity available under applicable law for approved products, as well as the potential for reduced time to market. Assuming positive clinical data, drugs intended to treat rare diseases or conditions also may qualify for fast track designation, breakthrough therapy designation, accelerated approval or priority review, any or all of which may speed the development and approval process and decrease drug development costs.
Establish Partnerships with Large Pharmaceutical Companies
We seek to establish strategic alliances and partnerships with larger pharmaceutical companies for the commercialization and co-development of our drug candidates.
In-License Unique Technology
We continually review opportunities to in-license and advance compounds in oncology that have value-creating potential and will strengthen our clinical development pipeline.
Capitalize on Our Management Team’s Expertise for Drug Development
Our management team possesses clinical development experience in oncology and several other therapeutic areas which facilitates strategic approaches to and competitive advantages in, the design, risk assessment and implementation of drug development programs. Our management team also has prior experience in pharmaceutical alliances, product launches and marketing.
Our Pipeline Drug Candidates
Clinical Stage Pipeline
RX-3117: Oral Small Molecule Nucleoside
RX-3117 is a novel, investigational, oral small molecule nucleoside compound. In pre-clinical models when activated (phosphorylated) by uridine-cytidine kinase 2, a protein that is overexpressed in various human cancer cells, RX-3117 is incorporated into DNA or RNA of cells and inhibits both DNA and RNA synthesis, which induces apoptotic cell death of tumor cells. We believe RX-3117 has therapeutic potential in a broad range of cancers including pancreatic, bladder, lung, cervical, non-small cell lung cancer and colon cancer. RX-3117 has received orphan drug designation from the FDA and the EC for the treatment of patients with pancreatic cancer. RX-3117 has also been shown in animal models to inhibit the growth of gemcitabine-resistant human cancers and improve overall survival.
RX-3117 has demonstrated broad spectrum anti-tumor activity against over 100 different human cancer cell lines and efficacy in 17 different mouse xenograft models. Notably, the efficacy of RX-3117 in the mouse xenograft models was superior to that of gemcitabine. Further, RX-3117 still retains its full anti-tumor activity in human cancer cell lines made resistant to the anti-tumor effects of gemcitabine. In August 2012, we reported the completion of an exploratory Phase 1 clinical trial of RX-3117 in cancer patients conducted in Europe to investigate the oral bioavailability, safety and tolerability of the compound. In this study, oral administration of a 50 mg dose of RX-3117 demonstrated an oral bioavailability of 56% and a plasma half-life (T1/2) of 14 hours. In addition, RX-3117 appeared to be safe and well tolerated in all subjects throughout the dose range tested.
Final results from the Phase 1b clinical trial of RX-3117 presented at the American Society of Clinical Oncology Annual Meeting in June 2016 showed evidence of single agent activity. Patients in the study had generally received four or more cancer therapies prior to enrollment. In this study, 12 patients experienced stable disease persisting for up to 276 days and three patients showed evidence of tumor burden reduction. A maximum tolerated dose of 700 mg was identified in the study. At the doses tested to date, RX-3117, administered orally, appeared to be safe and well tolerated with a predictable pharmacokinetic profile following oral administration.
In March 2016, we initiated a multi-center Phase 2a clinical trial of RX-3117 in patients with relapsed or refractory pancreatic cancer to further evaluate the safety and anti-cancer properties of this compound. Patients in the trial received a 700 mg daily oral dose of RX-3117, for five consecutive days, followed by two days off, for three weeks, followed by a week of rest, in a 28-day cycle for up to eight treatment cycles, or until their disease progressed. The study was designed as a two-stage study with 10 patients in stage 1 and an additional 40 patients in stage 2. According to pre-set criteria, if greater than 20% of the patients had an increase in progression free survival of more than four months, or an objective clinical response rate and reduction in tumor size, additional pancreatic cancer patients would be enrolled into stage 2. Secondary endpoints included time to disease progression, overall response rate and duration of response, as well as pharmacokinetic assessments and safety parameters.
In September 2016, we initiated stage 2 of this Phase 2a clinical trial based on the satisfaction of the predefined criteria for preliminary efficacy for stage 1 of the trial that showed RX-3117 was safe and well tolerated with preliminary efficacy in pancreatic cancer patients for whom three or more prior therapies had been ineffective. In January 2018, we presented the final data from this trial at the American Society of Clinical Oncology Gastrointestinal Cancers 2018 Annual Meeting. Encouraging progression free survival and evidence of tumor shrinkage was observed in patients with metastatic pancreatic cancer resistant to gemcitabine who had failed on multiple prior treatments.
In November 2017, we initiated a Phase 2a trial of RX-3117 in combination with Abraxane® in patients newly diagnosed with metastatic pancreatic cancer.
In September 2016, we commenced enrollment in a Phase 2a trial of RX-3117 in patients with advanced bladder cancer. This Phase 2a clinical trial is a multicenter, open-label, single-agent study of RX-3117 being conducted at 10 clinical centers in the United States. RX-3117 is being administered orally five times weekly on a three weeks on, one week off dosing schedule. The primary endpoint for the trial is an assessment of the progression free survival rate or an objective clinical response rate and reduction in tumor size. Secondary endpoints include time to disease progression, overall response rate and duration of response, as well as pharmacokinetic assessments and safety. In February 2018, we presented data from this trial at the American Society of Clinical Oncology Genitourinary Cancers 2018 Annual Meeting. Encouraging progression free survival and evidence of tumor shrinkage were observed in patients with advanced bladder cancer who had failed on multiple prior treatments including immunotherapy and gemcitabine.
Based on the progress of the RX-3117 clinical development program, we are continuing discussions with multiple companies to explore collaborative business structures in an effort to maximize the potential value of the program.
RX-5902 (Supinoxin): Potential First-in-Class Inhibitor of Phosphorylated p68
RX-5902 is a potential first-in-class small molecule inhibitor of the interaction between phosphorylated-p68, a protein that we believe plays a key role in cancer growth, progression and metastasis and beta-catenin. Many cancers are driven by beta-catenin-mediated gene expression. Phosphorylated p68, which is highly expressed in cancer cells, but not in normal cells, results in up-regulation of cancer-related genes and a subsequent proliferation of cancer cells and tumor growth. RX-5902 selectively blocks the phosphorylated p68-beta catenin interaction, thereby decreasing the proliferation or growth of cancer cells. In pre-clinical tissue culture models and in-vivo xenograft models, RX-5902 has exhibited single-agent tumor growth inhibition, potential synergy with cytotoxic agents and activity against drug resistant cancer cells. In particular, in in-vivo xenograft models of human triple negative breast cancer and pancreatic cancer, treatment with RX-5902 on days one through 20 in mouse models produced a dose-dependent inhibition of tumor growth and a survival benefit.
RX-5902 was evaluated in a Phase 1 dose-escalation clinical trial in cancer patients with solid tumors designed to evaluate the safety, tolerability, dose-limiting toxicities and the recommended Phase 2 dose. Secondary endpoints include pharmacokinetic analyses and an evaluation of the preliminary anti-tumor effects of RX-5902. We completed enrollment in this study in 2016.
Updated results from the Phase 1 clinical trial of RX-5902 were presented in October 2016 at the 2016 European Society for Medical Oncology Congress.
The results showed evidence of single-agent, clinical activity of RX-5902. In this study, RX-5902 preliminarily appeared to be safe and well tolerated at the doses and dosing schedules tested with no dose limiting toxicities or treatment-related serious adverse events. The most frequently reported drug related adverse events were mild nausea, vomiting and fatigue. Initial signs of clinical activity have been observed. Twenty-four subjects were enrolled (11 female, 13 male), and seven subjects experienced stable disease in breast, neuroendocrine, paraganglioma, head/neck or colorectal cancer. Three subjects received treatment for more than one year. Approximately 55% of the subjects had received four or more therapies prior to their enrollment in the Phase 1 clinical study.
We initiated a Phase 2a study of RX-5902 in patients with triple negative breast cancer in February 2017. The study will evaluate the safety and preliminary efficacy of RX-5902 in patients with metastatic triple negative breast cancer who have failed prior treatments. We also plan to evaluate RX-5902 in combination with other anticancer agents in TNBC, assuming positive data from this initial study.
Based on the progress of the RX-5902 clinical development program, we are continuing our discussions with multiple companies to explore collaborative business structures in an effort to maximize the potential commercial value of the program.
RX-0201 (Archexin): Potential Best-in-Class Anti-Cancer Akt-1 Inhibitor
RX-0201 is a potential best-in-class, potent anti-sense inhibitor of protein kinase Akt-1 synthesis and activity, which we believe plays a critical role in cancer cell proliferation, survival, angiogenesis, metastasis and drug resistance. RX-0201 has received orphan drug designation from the FDA for RCC, glioblastoma, ovarian cancer, stomach cancer and pancreatic cancer. We believe RX-0201 is differentiated from other Akt-1 inhibitors by its ability to inhibit both activated and inactivated forms of Akt-1, and as a result it is not expected to lead to drug resistance, which has been observed with other protein kinase inhibitors. Other targeted drugs may only inhibit inactivated Akt-1 and may also cause drug resistance. Akt-1 is over-activated in patients with many cancers, including breast, colorectal, gastric, pancreatic, prostate and melanoma cancers. Akt-1 activity may be inhibited by signaling molecules upstream of Akt-1 in cancer cells through the use of vascular endothelial growth factor and epidermal growth factor receptor inhibitors, but this treatment only indirectly affects the activity of native Akt-1. Because signal transmission for cancer progression and resistance occurs when Akt-1 is activated, we believe it is also important to inhibit activated Akt-1. We believe RX-0201 inhibits both activated and native Akt-1.
RX-0201 is an antisense oligonucleotide compound that is complementary to Akt-1 mRNA and highly selective for inhibiting mRNA expression, which leads to reduced production of Akt-1 protein. RX-0201 preliminarily appeared to be safe and well tolerated with minimal side effects in a Phase 1 study in patients with advanced cancers, where Grade 3 fatigue was the only dose-limiting toxicity and no significant hematological abnormalities were observed. A nano-liposomal formulation of RX-0201 is being developed under a collaboration with Haichang using Haichang’s proprietary QTzomes™ technology. Under the agreement, Haichang intends to conduct a Phase 2a proof-of-concept clinical study in HCC in China.
We completed a Phase 2a clinical trial for RX-0201 that was designed to assess the safety and efficacy of RX-0201 in combination with gemcitabine. RX-0201 was shown to be safe and well tolerated with a preliminary indication of activity.
In January 2014, we initiated a Phase 2a proof-of-concept clinical trial of RX-0201 to study its safety and efficacy in combination with Afinitor® (everolimus) in patients with metastatic RCC. The trial is being conducted in two stages. Stage 1 was a dose ranging study, with up to three dose groups with three RCC patients each, to determine its maximum tolerated dose (“MTD”) in combination with everolimus. In January 2016, we completed Stage 1 of the study and commenced enrollment in Stage 2, which is a randomized, open-label, two-arm dose expansion study of everolimus versus RX-0201 in combination with everolimus to determine safety and efficacy of the combination. This phase of the trial (Stage 2) was anticipated to enroll up to 40 RCC patients randomized to receive either RX-0201 in combination with everolimus, or everolimus alone, in a ratio of 2:1 The MTD was determined to be 250 mg/m2/day of RX-0201, which was identified in Stage 1 and was administered in Stage 2 along with 10 mg of everolimus compared to 10 mg everolimus alone. In February 2018, following a portfolio review of assets and in response to the changing treatment landscape for RCC patients over the past two years with the approval of new therapies by the FDA, we announced that we are winding down internally funded programs of RX-0201 including the cessation of enrollment in this trial. Patients currently enrolled in the trial will continue to be followed.
Research and Development Process
We have engaged third-party contract research organizations and other investigators and collaborators, such as universities and medical institutions, to conduct our pre-clinical studies, toxicology studies and clinical trials. Engaging third-party contract research organizations is typical practice in our industry. However, relying on such organizations means that the clinical trials and other studies described above are being conducted at external locations and that the completion of these trials and studies is not within our direct control. Trials and studies may be delayed due to circumstances outside our control, and such delays may result in additional expenses for us.
Competition
We compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, as well as academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as more experience in:
| · |
developing drugs;
|
| · |
undertaking pre-clinical testing and human clinical trials;
|
| · |
obtaining FDA and other regulatory approvals of drugs;
|
| · |
formulating and manufacturing drugs; and
|
| · |
launching, marketing and selling drugs.
|
Large pharmaceutical companies currently sell both generic and proprietary compounds for the treatment of cancer. In addition, companies developing oncology therapies represent substantial competition. Many of these organizations have substantially greater capital resources, larger research and development staff and facilities, history in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to attract qualified personnel, parties for acquisitions, joint ventures or other collaborations.
We are aware of products under development by our competitors that target the same indications as our clinical stage drug candidates. If approved, RX-3117 could compete with other compounds with an anti-metabolite mechanism of action in cancers, such as NUC-1301 (Acelarin®), which is under development by NuCana and other approved nucleoside analogues such as capecitabine and gemcitabine. We are not currently aware of known inhibitors of phosphorylated p68 that would compete with RX-5902 if RX-5902 were approved, but other drugs with a different mechanism of action are in development for the same indications, such as Immunomedics’ sacitazumab govitecan and Celldex’s glembatumumab vedotin, both in development for triple negative breast cancer. Our competitors may succeed in developing products that are safer and/or more effective than ours, which could render our product candidates less competitive prior to recovery by us of expenses incurred with respect to their development.
Government Regulation
Regulation by governmental authorities in the United States and in other countries is a significant consideration in our product development, manufacturing and, upon approval of our product candidates, marketing strategies. We expect that all our drug candidates will require regulatory approval by the FDA and by similar regulatory authorities in foreign countries prior to commercialization and will be subjected to rigorous pre-clinical, clinical, and post-approval testing to demonstrate safety and effectiveness, as well as other significant regulatory requirements and restrictions in each jurisdiction in which we would seek to market our products. U.S. federal laws and regulations govern the testing, development, manufacture, quality control, safety, effectiveness, approval, storage, labeling, record keeping, reporting, distribution, import, export and marketing of all biopharmaceutical products intended for therapeutic purposes. We believe that we and the third parties that work with us are in compliance in all material respects with currently applicable rules and regulations, however, any failure to comply could have a material negative impact on our ability to successfully develop and commercialize our products, and therefore on our financial performance. In addition, these rules and regulations are subject to change. For example, in December 2016, the 21st Century Cures Act (the “Cures Act”) was signed into law. The Cures Act included numerous provisions that may be relevant to our product candidates, including provisions designed to speed development of innovative therapies and provide funding for certain cancer-related research and technology development. Because the Cures Act is still relatively new, it is difficult to foresee whether, how, or when it may affect our business. Further legislative and regulatory changes appear possible in the 115th United States Congress and under the Trump Administration, and it is difficult to foresee whether, how, or when such changes may affect our business.
Obtaining governmental approvals and maintaining ongoing compliance with applicable regulations are expected to require the expenditure of significant financial and human resources not currently at our disposal. We plan to fulfill our short-term needs through consulting agreements and joint ventures with academic or corporate partners while developing our own internal infrastructure for long-term corporate growth.
Development and Approval
The process to obtain approval for biopharmaceutical compounds for commercialization in the United States and many other countries is lengthy, complex and expensive, and the outcome is far from certain. Although foreign requirements for conducting clinical trials and obtaining approval may be different than in the United States, they often are equally rigorous and the outcome cannot be predicted with confidence. A key component of any submission for approval in any jurisdiction is pre-clinical and clinical data demonstrating the product’s safety and effectiveness.
Pre-clinical Testing. Before testing any compound in humans in the United States, a company must develop pre-clinical data, generally including laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological studies in animal species to assess safety and quality. Certain types of animal studies must be conducted in compliance with the FDA’s Good Laboratory Practice regulations and the Animal Welfare Act, which is enforced by the Department of Agriculture.
IND Application. In the United States, FDA regulations require that the person or entity sponsoring or conducting a clinical study for the purpose of investigating a candidate’s safety and effectiveness submit to the FDA an investigational new drug (“IND”) application, which contains pre-clinical testing results and provides a basis for the FDA to conclude that there is an adequate basis for testing the drug in humans. If the FDA does not object to the IND application within 30 days of submission, the clinical testing proposed in the IND may begin. Even after the IND has gone into effect and clinical testing has begun, the FDA may put the clinical trials on “clinical hold,” suspending (or in some cases, ending) them because of safety concerns or for other reasons.
Clinical Trials. Clinical trials involve administering a drug to human volunteers or patients under the supervision of a qualified clinical investigator. Clinical trials are subject to extensive regulation. In the United States, this includes compliance with the FDA’s bioresearch monitoring regulations and Good Clinical Practice (“GCP”) requirements, which establish standards for conducting, recording data from, and reporting the results of, clinical trials, with the goal of assuring that the data and results are credible and accurate and that study participants’ rights, safety and well-being are protected. Each clinical trial must be conducted under a protocol that details the study objectives, parameters for monitoring safety and the efficacy criteria, if any, to be evaluated. The protocol is submitted to the FDA as part of the IND and reviewed by the agency before the study begins. Additionally, each clinical trial must be reviewed, approved and conducted under the auspices of an Institutional Review Board (“IRB”). The sponsor of a clinical trial, the investigators and IRBs each must comply with requirements and restrictions that govern, among other things, obtaining informed consent from each study subject, complying with the protocol and investigational plan, adequately monitoring the clinical trial, and timely reporting adverse events. Foreign studies conducted under an IND must meet the same requirements applicable to studies conducted in the United States. However, if a foreign study is not conducted under an IND, the data may still be submitted to the FDA in support of a product application, if the study was conducted in accordance with GCP and the FDA is able to validate the data.
Sponsors of clinical trials are required to make public certain information about active clinical trials and trial results by posting the information on government or independent websites, such as http://clinicaltrials.gov. Clinical testing is typically performed in three phases.
In Phase 1, the drug is administered to a small number of human subjects to confirm its safety and to develop detailed profiles of its pharmacological and pharmacokinetic actions (i.e., absorption, distribution, metabolism, and excretion). Although Phase 1 trials typically are conducted in healthy human subjects, in some instances (including, for example, with some cancer therapies) the study subjects are patients with the targeted disease or condition.
In Phase 2, the drug is administered to groups of patients (usually no more than several hundred) to develop initial data regarding efficacy against the targeted disease and determine the requisite dose and dose intervals, and generate additional information regarding the drug’s safety. In a typical development program, additional animal toxicology studies precede this phase. In some cases, the trial can be split into Phase 2a and 2b studies in order to test smaller subject pools. Some Phase 1 clinical studies may proceed in parallel with some Phase 2 studies.
In Phase 3, the drug is administered to a larger group of patients (usually from several hundred to several thousand or more). Phase 3 studies also may include patients with concomitant diseases and medications. Larger patient populations are evaluated in Phase 3 at multiple study sites and registration studies may be conducted concurrently for the sake of time and efficiency. The extensive clinical testing is intended to obtain additional information about product safety and effectiveness necessary to evaluate the drug’s overall risk-benefit profile and to provide a basis for physician labeling. Phase 3 data often form the core basis on which the FDA evaluates the product’s safety and effectiveness when considering an application to market the drug.
The study sponsor, the FDA or an IRB may suspend or terminate a clinical trial at any time on various grounds, including a determination that study subjects are being exposed to an unacceptable health risk. Additionally, success in early-stage clinical trials does not assure success in later-stage clinical trials, and data from clinical trials are not always conclusive and may be subject to alternative interpretations that could delay, limit or prevent approval.
NDA Submission and Review. After completing the clinical studies, a sponsor seeking approval to market a drug in the United States submits to the FDA a New Drug Application (“NDA”). The NDA is a comprehensive, multi-volume application intended to demonstrate the product’s safety and effectiveness and includes, among other things, pre-clinical and clinical data, information about the drug’s composition, the sponsor’s plans for manufacturing and packaging and proposed labeling. When an NDA is submitted, the FDA makes an initial determination as to whether the application is sufficiently complete to be accepted for review. If the application is not, the FDA may refuse to accept the NDA for filing and request additional information. A refusal to file, which requires resubmission of the NDA with the requested additional information, delays review of the application.
FDA performance goals regarding the timeliness of NDA review generally provide for action on an NDA within 12 months of its submission. That deadline can be extended under certain circumstances, including by the FDA’s requests for additional information. The targeted action date can also be shortened to eight months after submission for products that are granted priority review designation because they are intended to treat serious or life-threatening conditions and demonstrate the potential to address unmet medical needs. Additionally, the FDA has programs for enhanced communication and consultation and other steps to expedite development and review of such products. For example, the Fast Track program is intended to facilitate the development and review of new drugs that demonstrate the potential to address unmet medical needs involving serious or life-threatening diseases or conditions. If a drug receives Fast Track designation, the FDA may review sections of the NDA on a rolling basis, rather than requiring the entire application to be submitted to begin the review. Products with Fast Track designation also may be eligible for more frequent meetings and correspondence with the FDA about the product’s development. Other FDA programs intended to expedite development and review include Accelerated Approval, which allows approval on the basis of a surrogate endpoint that is reasonably likely to predict clinical benefit and Breakthrough Therapy designation, which is available for drugs under development for serious or life-threatening conditions and where preliminary clinical evidence shows that the drug may have substantial improvement on at least one clinically significant endpoint over available therapy. If a drug receives Breakthrough Therapy designation, it will be eligible for all of the benefits of Fast Track designation, as well as for more intensive guidance from the FDA on an efficient drug development program and a commitment from the agency to involve senior FDA managers in such guidance. Even if a product qualifies for Fast Track designation or Breakthrough Therapy designation, the FDA may later decide that the product no longer meets the conditions for designation, and/or may determine that the product does not meet the standards for approval. We anticipate, but cannot ensure, that our product candidates will qualify for such programs.
If it concludes that an NDA does not meet the regulatory standards for approval, the FDA typically issues a Complete Response letter, which communicates the reasons for the agency’s decision not to approve the application and may request additional information, including additional clinical data. An NDA may be resubmitted with the deficiencies addressed, but that does not guarantee approval. Data from clinical trials are not always conclusive, and the FDA’s interpretation of data may differ from the sponsor’s. Obtaining approval can take years, requires substantial resources and depends on a number of factors, including the severity of the targeted disease or condition, the availability of alternative treatments, and the risks and benefits demonstrated in clinical trials. Additionally, as a condition of approval, the FDA may impose restrictions that could affect the commercial prospects of a product, such as a Risk Evaluation and Mitigation Strategy, and could require post-approval commitments to conduct additional studies or conduct surveillance programs to monitor the drug’s effects.
Moreover, once a product is approved, information about its safety or effectiveness from broader clinical use may limit or prevent successful commercialization, either because of regulatory action or market forces. Post-approval modifications to a drug product, such as changes in indications, labeling or manufacturing processes or facilities, may require development and submission of additional information or data in a new or supplemental NDA, which would also require FDA approval.
One of our drug candidates, RX-0201 is an antisense oligonucleotide (“ASO”) compound. To date, the FDA has not approved any NDAs for any ASO compounds for cancer treatment; however, the FDA has approved the ASO compounds fomivirsen (marketed as Vitravene®) as a treatment for cytomegalovirus retinitis, and mipomersen sodium (marketed as Kynamro®), as a treatment for homozygous familial hypercholesterolemia. In addition, RX-0201 is in a drug class known as Akt-1 inhibitors, and drugs from this class have not been approved by the FDA to date.
We have not submitted an NDA for any of our drug candidates.
Exclusivity and Patent Protection. In the United States and elsewhere, certain regulatory exclusivities and patent rights can provide an approved drug product with protection from certain competitors’ products for a period of time and within a certain scope. In the United States, those protections include exclusivity under the Orphan Drug Act, which is available for drugs intended to treat rare diseases or conditions, which generally are diseases or conditions that affect fewer than 200,000 persons in the United States. If a sponsor demonstrates that a drug is intended to treat a rare disease or condition and meets other qualifying criteria, the FDA grants orphan drug designation to the product for that use. A product that has received orphan drug designation is eligible for research and development tax credits and is exempt from user fees under certain circumstances. Additionally, a drug that is approved for its orphan-designated indication generally receives seven years of orphan drug exclusivity. During that period, the FDA generally may not approve any other application for a product containing the same active moiety and proposed for the same indication. There are exceptions, however, most notably when the later product is shown to be clinically superior to the product with exclusivity. An approved orphan drug also may qualify for an exemption from the branded prescription drug fee. Products that qualify for orphan designation may also qualify for other FDA programs that are intended to expedite the development and approval process and, as a practical matter, clinical trials for orphan products may be smaller, simply because of the smaller patient population. Nonetheless, the same approval standards apply to orphan-designated products as for other drugs.
RX-0201 has received orphan drug designation from the FDA for RCC, glioblastoma, ovarian cancer, stomach cancer and pancreatic cancer. RX-3117 received orphan drug designation for pancreatic cancer from the FDA in September 2014.
A medicinal product may be granted an orphan designation in the EU if: (i) it would be used to treat or prevent a life-threatening or chronically debilitating condition and either affects no more than five in 10,000 people in the EU or for economic reasons would be unlikely to be developed without incentives; and (ii) no satisfactory method of diagnosis, prevention or treatment of the condition concerned exists, or, if such a method exists, the medicinal product would be of significant benefit to those affected by the condition. The application for orphan designation must be submitted to the European Medicines Agency (“EMA”) and approved prior to market authorization. Once authorized, orphan medicinal products are entitled to ten years of market exclusivity. During this ten-year period, with limited exceptions, neither the competent authorities of the EU Member States, the EMA, nor the EC are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. However, marketing authorization may be granted to a similar medicinal product with the same orphan indication during that period with the consent of the holder of the marketing authorization or if the manufacturer of the product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if the latter product is safer, more effective or otherwise clinically superior to the original product. The period of market exclusivity may be reduced to six years if it can be demonstrated on the basis of available evidence that the original orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity.
RX-3117 received orphan designation from the EC in January 2018.
Generic Competition. Any drug candidates approved for commercial marketing under an NDA would be subject to the provisions of the Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act. Among other things, the Hatch-Waxman Act establishes two abbreviated approval pathways for drug products that are in some way follow-on versions of already approved NDA products, including generic versions of the approved product, which may be approved under an Abbreviated New Drug Application by a showing that the generic product is the “same as” the approved product in key respects. Those abbreviated approval pathways generally are available, however, after expiration of certain periods of regulatory exclusivity and/or extended patent protection for the approved NDA product, which the Hatch-Waxman Act also provides. These protections include: (1) five years of regulatory exclusivity for a new chemical entity (generally, the first approval of a product containing a particular active moiety), during which an application for a follow-on product cannot be reviewed; (2) three years of exclusivity for the approval of an NDA or supplemental NDA that contains data from new clinical investigations that were necessary for approval, during which the follow-on product may not receive final approval; and (3) up to five years’ extension of the term of a patent covering a drug that contains an active ingredient not previously approved. The Hatch-Waxman Act also provides a means for the sponsor of an approved NDA to act before approval of a proposed abbreviated NDA to sue to protect patents claiming the drug substance, drug product, or an approved method of using the drug. The laws of other key markets likewise create both opportunities for exclusivity periods and patent protections and the possibility of generic competition once such periods or protections have either expired or have been successfully challenged by generic entrants.
Post-Approval Regulation
Once approved, products are subject to continuing extensive regulation by the FDA. If ongoing regulatory requirements are not met, or if safety problems occur after a product reaches market, the FDA may take actions to change the conditions under which the product is marketed, including suspending or even withdrawing approval. In addition to FDA regulation, the healthcare industry, and therefore our business, is also subject to extensive federal, state, local and foreign regulation.
Good Manufacturing Practices. Companies engaged in manufacturing drug products or their components must comply with applicable current Good Manufacturing Practice (“cGMP”) requirements, which include requirements regarding organization and training of personnel, building and facilities, equipment, control of components and drug product containers, closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls and records and reports. The FDA inspects equipment, facilities and manufacturing processes before approval and conducts periodic re-inspections after approval. Failure to comply with applicable cGMP requirements or the conditions of the product’s approval may lead the FDA to take administrative enforcement action. Although we periodically monitor FDA compliance of the third parties on which we rely for manufacturing our drug products, we cannot be certain that our present or future third-party manufacturers will consistently comply with cGMP or other applicable FDA regulatory requirements.
Sales and Marketing. Once a product is approved, its advertising, promotion and marketing will be subject to close regulation, including with regard to promotion to healthcare practitioners, direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities and promotional activities involving the internet. In addition to FDA restrictions on marketing of pharmaceutical products, state and federal fraud and abuse laws have been applied to restrict certain marketing practices in the pharmaceutical industry for many years. Some of the pertinent laws are open to a variety of interpretations. In addition, these laws and their interpretations are subject to change.
Other Requirements. Companies that manufacture or distribute drug products pursuant to approved NDAs must meet numerous other regulatory requirements, including adverse event reporting, submission of periodic reports, and record-keeping obligations.
Fraud and Abuse Laws. At such time as we market, sell and distribute any products for which we obtain marketing approval, it is possible that our business activities could be subject to scrutiny and enforcement under one or more federal or state health care fraud and abuse laws and regulations, which could affect our ability to operate our business. These restrictions under applicable federal and state health care fraud and abuse laws and regulations that may affect our ability to operate include:
| · |
The federal Anti-Kickback Law, which prohibits, among other things, knowingly or willingly offering, paying, soliciting or receiving remuneration, directly or indirectly, in cash or in kind, to induce or reward the purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any health care items or service for which payment may be made, in whole or in part, by federal healthcare programs such as Medicare and Medicaid. This statute has been interpreted to apply to arrangements between pharmaceutical companies on one hand and prescribers, purchasers and formulary managers on the other. Further, the Affordable Care Act clarified among other things that liability may be established under the federal Anti-Kickback Law without proving actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Law constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Although there are a number of statutory exemptions and regulatory safe harbors to the federal Anti-Kickback Law protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exemption or safe harbor, or for which no exception or safe harbor is available, may be subject to scrutiny;
|
| · |
The federal civil False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the federal government. Many pharmaceutical and other healthcare companies have been investigated and have reached substantial financial settlements with the federal government under the civil False Claims Act for a variety of alleged improper marketing activities, including: providing free product to customers with the expectation that the customers would bill federal programs for the product; providing sham consulting fees, grants, free travel and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug payment rates under government healthcare programs. In addition, in recent years the government has pursued civil False Claims Act cases against a number of pharmaceutical companies for causing false claims to be submitted as a result of the marketing of their products for unapproved, and thus non-reimbursable, uses. Pharmaceutical and other healthcare companies also are subject to other federal false claim laws, including, among others, federal criminal healthcare fraud and false statement statutes that extend to non-government health benefit programs;
|
| · |
Analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to items or services reimbursed under Medicaid and other state programs or, in several states, apply regardless of the payor. Some state laws also require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to certain healthcare providers in the states. Other states prohibit providing meals to prescribers or other marketing related activities. In addition, California, Connecticut, Nevada and Massachusetts require pharmaceutical companies to implement compliance programs or marketing codes of conduct.
|
| · |
The federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, requires certain pharmaceutical manufacturers to engage in extensive tracking of payments and other transfers of value to physicians and teaching hospitals, and to submit such data to the Centers for Medicare and Medicaid Services (“CMS”), which will then make all of this data publicly available on the CMS website. Pharmaceutical manufacturers with products for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program are required to track reportable payments and must submit a report to CMS on or before the 90th day of each calendar year disclosing reportable payments made in the previous calendar year. Failure to comply with the reporting obligations may result in civil monetary penalties;
|
| · |
The federal Foreign Corrupt Practices Act of 1997 and other similar anti-bribery laws in other jurisdictions generally prohibit companies and their intermediaries from providing money or anything of value to officials of foreign governments, foreign political parties, or international organizations with the intent to obtain or retain business or seek a business advantage. Recently, there has been a substantial increase in anti-bribery law enforcement activity by U.S. regulators, with more frequent and aggressive investigations and enforcement proceedings by both the Department of Justice and the U.S. Securities and Exchange Commission (the “SEC”). Violations of United States or foreign laws or regulations could result in the imposition of substantial fines, interruptions of business, loss of supplier, vendor or other third-party relationships, termination of necessary licenses and permits and other legal or equitable sanctions. Other internal or government investigations or legal or regulatory proceedings, including lawsuits brought by private litigants, may also follow as a consequence.
|
Violations of any of the laws described above or any other governmental regulations are punishable by significant civil, criminal and administrative penalties, damages, fines and exclusion from government-funded healthcare programs, such as Medicare and Medicaid. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
Privacy Laws. We are also subject to laws and regulations covering data privacy and the protection of health-related and other personal information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business, including recently enacted laws in all jurisdictions where we operate. Numerous federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use and disclosure of personal information. Failure to comply with such laws and regulations could result in government enforcement actions and create liability for us (including the imposition of significant penalties), private litigation and/or adverse publicity that could negatively affect our business. In addition, if we successfully commercialize our drug candidates, we may obtain patient health information from healthcare providers who prescribe our products and research institutions we collaborate with, and they are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (“HIPAA”). Although we are not directly subject to HIPAA other than with respect to providing certain employee benefits, we could potentially be subject to criminal penalties if we knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug candidates for which we may obtain regulatory approval. The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates even if our product candidates obtain marketing approval.
Our ability to commercialize any products successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available in a timely manner from government third-party payors, including government healthcare programs such as Medicare and Medicaid, commercial health insurers and managed care organizations. Government authorities and other third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. Third-party payors may limit coverage to specific products on an approved list, or formulary, which may not include all of the FDA-approved products for a particular indication. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government healthcare programs and other third-party payors are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy, and have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available promptly or at all for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases. Limited coverage may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any product candidate for which we obtain marketing approval.
Payors also are increasingly considering new metrics as the basis for reimbursement rates, such as average sales price (“ASP”), average manufacturer price (“AMP”) and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS surveys and publishes retail community pharmacy acquisition cost information in the form of National Average Drug Acquisition Cost (“NADAC”) files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. It may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payors to cover our products for which we receive regulatory approval.
If we successfully commercialize any of our products, we may participate in the Medicaid Drug Rebate Program. Participation is required for federal funds to be available for our products under Medicaid and Medicare Part B. Under the Medicaid Drug Rebate Program, we would be required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Part B of the Medicare program.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients.
In addition, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by the Department of Veterans Affairs (the “VA”), Department of Defense (“DoD”), Public Health Service, and Coast Guard (the “Big Four agencies”) and certain federal grantees, a manufacturer also must participate in the VA Federal Supply Schedule (“FSS”) pricing program, established by Section 603 of the Veterans Health Care Act of 1992 (the “VHCA”). Under this program, the manufacturer is obligated to make its covered drugs (innovator multiple source drugs, single source drugs, and biologics) available for procurement on an FSS contract and charge a price to the Big Four agencies that is no higher than the Federal Ceiling Price (“FCP”), which is a price calculated pursuant to a statutory formula. The FCP is derived from a calculated price point called the “non-federal average manufacturer price” (“Non-FAMP”), which we will be required to calculate and report to the VA on a quarterly and annual basis. Moreover, pursuant to Defense Health Agency (“DHA”) regulations, manufacturers must provide rebates on utilization of their innovator and single source products that are dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. The formula for determining the rebate is established in the regulations and is based on the difference between the annual non-federal average manufacturer price and the Federal Ceiling Price, each required to be calculated by us under the VHCA. The requirements under the 340B, FSS, and TRICARE programs could reduce the revenue we may generate from any products that are commercialized in the future and could adversely affect our business and operating results.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Limited coverage may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. Third-party payors also may seek additional clinical evidence, including expensive pharmacoeconomic studies, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations, before covering our products for those patients. If reimbursement is available only for limited indications, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
United States Healthcare Reform
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidate for which we obtain marketing approval. The United States government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
In recent years, Congress has considered reductions in Medicare reimbursement levels for drugs administered by physicians. CMS, the agency that administers the Medicare and Medicaid programs, also has authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products, which in turn would affect the price we can receive for those products. While Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Affordable Care Act. This law substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum Medicaid rebate for both branded and generic drugs, expanded the 340B program, and revised the definition of AMP, which could increase the amount of Medicaid drug rebates manufacturers are required to pay to states. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service Medicaid utilization, to include the utilization of Medicaid managed care organizations as well and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the amount of rebates due on those drugs. On February 1, 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016.
Moreover, certain legislative changes to and regulatory changes under the Affordable Care Act have occurred in the 115th United States Congress and under the Trump Administration. For example, the Tax Cuts and Jobs Act, enacted on December 22, 2017, eliminated the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, commonly referred to as the individual mandate, beginning in 2019. Additional legislative changes to and regulatory changes under the Affordable Care Act remain possible. Any such changes could decrease the number of individuals with health coverage. We expect that the Affordable Care Act, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to successfully commercialize our product candidates, if approved.
The Affordable Care Act requires pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government. Each individual pharmaceutical manufacturer pays a prorated share of the branded prescription drug fee of $4.1 billion in 2018 and $2.8 billion in years thereafter, based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. Furthermore, the Affordable Care Act requires manufacturers to provide a 50% discount off the negotiated price of prescriptions filled by beneficiaries in the Medicare Part D coverage gap, referred to as the “donut hole.” The Bipartisan Budget Act of 2018 increased the required manufacturer discount to 70% off the negotiated price for Medicare Part D beneficiaries in the donut hole beginning in 2019.
The Affordable Care Act also expanded the Public Health Service’s 340B drug pricing discount program. As noted above, the 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. The Affordable Care Act expanded the 340B program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by the Affordable Care Act. The Affordable Care Act exempts “orphan drugs”—those designated under section 526 of the Food, Drug, and Cosmetic Act—from the ceiling price requirements for these newly-eligible entities. Because the 340B ceiling price is determined based on AMP and Medicaid drug rebate data, the revisions to the Medicaid rebate formula and AMP definition could cause the required 340B discounts to increase.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. For example, recent legislative enactments have resulted in Medicare payments being subject to a two percent reduction, referred to as sequestration, until 2027. Continuation of sequestration or enactment of other reductions in Medicare reimbursement for drugs could affect our ability to achieve a profit on any candidate products that are approved for marketing.
We expect that the Affordable Care Act, as well as other healthcare reform measures that have been adopted and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product and could seriously harm our future revenues. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a number of significant regulations in other jurisdictions regarding clinical trials, approval, manufacturing, marketing and promotion and safety reporting. These requirements and restrictions vary from country to country, but in many instances are similar to the United States requirements, and failure to comply with them could have the same negative effects as noncompliance in the United States.
Sales and Marketing
We do not currently have the sales and marketing infrastructure in place that would be necessary to sell and market products. As our drug candidates progress in clinical trials, we may build the commercial infrastructure that would be needed to successfully market and sell any successful drug candidate. For drug candidates that may require larger clinical trials or sales efforts, we intend to establish strategic alliances and partnerships with large pharmaceutical companies during the development process.
Research Technologies
Our research technologies are focused on our proprietary multi-target aimed ligands platform and nano-based drug delivery. For example, we have developed unique proprietary drug delivery nano-systems that we believe may increase the availability of a drug at the disease site, minimize adverse reactions, and provide longer duration of action. We are currently testing multiple nanoliposomal- and nanopolymer-based anti-cancer drugs.
In some circumstances, we partner with universities, research institutions and other organizations to obtain research and development services. For a discussion of collaboration arrangements pursuant to which we obtain research and development services, see “Collaboration and License Agreements” below in this Item 1.
Manufacturing and Distribution
We have no experience in drug formulation or manufacturing, and we lack the resources and expertise to formulate or manufacture our own drug candidates internally. Therefore, we rely on third-party expertise to support us in this area. We have entered into contracts with third-party manufacturers to manufacture, supply, store and distribute supplies of our drug candidates for our clinical trials. If any of our drug candidates receive FDA approval, we expect to rely on third-party contractors to manufacture our drugs. We have no current plans to build internal manufacturing capacity for any product, and we have no long-term supply arrangements.
Intellectual Property
We generally seek proprietary patent and intellectual property (“IP”) protection for our drug candidates, processes, and other know-how. In addition to patent protection, we rely upon trade secrets, know-how, continuing technological innovation and licensing opportunities to develop and safeguard and maintain our IP.
We hold U.S. and foreign patents for our drug candidates that expire from 2023 to 2036. We hold U.S., European and Japanese patents for RX-3117, RX-5902 and RX-0201. In addition to these patents, we have issued or pending patents in other jurisdictions.
The patent portfolios for our most advanced programs are summarized below:
RX-3117:
The RX-3117 patent portfolio consists of three patent families. The first family consists of patents that have been issued in the United States, Europe, Japan and other jurisdictions. The patents in this family include composition of matter, use, and process claims of varying scope, including picture claims to RX-3117 or a pharmaceutically acceptable salt thereof. The patents in this first family expire in 2025 but may be extended by patent term extension and orphan and market exclusivity. The second family consists of patents that have been issued in the United States, and are pending in Europe, Japan and other jurisdictions. The patents in the second family include process claims that cover RX-3117. The patents in this second family expire in 2034. The third family consists of a patent that is issued in the United States and pending in other jurisdictions. This patent includes use claims that cover the administration of RX-3117. This patent expires in 2036.
RX-5902 (Supinoxin):
The RX-5902 patent portfolio consists of three patent families. The first family consists of patents that have been issued in the United States and Europe and are pending in Japan and other jurisdictions. The patents in the first family include composition of matter, use, and process claims of varying scope, including picture claims to RX-5902 or a pharmaceutically acceptable salt thereof. The patents in this first family expire in 2025 and may be extended up to five years in the United States. We also expect RX-5902 will be protected with market exclusivity in Europe for a minimum of ten years post-approval and in Japan for eight years. The second family consists of patents that are issued in the United States and Japan and pending in Europe and other jurisdictions. The patents in the second family include composition of matter and process claims that cover RX-5902. The patents in this second family would expire in 2034. The third family consists of a patent that is issued in the United States and pending elsewhere. The patent in the third family includes use claims that cover RX-5902. This patent will expire in 2036.
RX-0201 (Archexin):
The RX-0201 patent portfolio consists of a patent family that includes patents that have been issued in the United States, Europe, Japan and other jurisdictions. The patents in this family include composition of matter and use claims of varying scope, including picture claims to RX-0201 or a pharmaceutically acceptable salt thereof. The expiration date of these patents ranges from 2023 to 2025, and may be extended by up to five years in certain countries including the United States. In addition, it is expected that RX-0201 will be protected from generic launches by market and orphan designations for up to seven years in the United States, and ten years in Europe and Japan.
Collaboration and License Arrangements
We have numerous collaborative research and development relationships with universities, research institutions pharmaceutical companies and other organizations.
Zhejiang Haichang Biotechnology Co., Ltd.
In February 2018, we entered into a research and development collaboration agreement with Haichang, a privately owned specialized biotechnology company incorporated in Hangzhou, China and focused on the development and manufacture of complex intravenous pharmaceutical products primarily for cancer treatment. Under the agreement, Haichang will develop a nano-liposomal formulation of RX-0201 using its proprietary QTzomes™ technology and will conduct certain pre-clinical and clinical activities through completion of a Phase 2a proof-of-concept clinical trial in HCC in China. Haichang will fund all development activities through completion of the Phase 2a clinical trial up to an aggregate amount of $10,000,000 and the parties will share downstream licensing fees and royalties paid by third parties in an agreed ratio in connection with the further development and commercialization of RX-0201 for the treatment of HCC. If Haichang exercises its right of first negotiation after completion of the Phase 2a clinical trial to obtain an exclusive license to further develop and commercialize RX-0201 in China, Haichang will pay customary license fees, milestone payments and royalties to Rexahn. Any clinical trials conducted by Haichang will be designed to meet both FDA and China Food and Drug Administration requirements.
Rexgene Biotech Co., Ltd. (“Rexgene”) and Next-BT Co. Ltd. (“Next-BT”)
In February 2003, we entered into a research collaboration agreement with Rexgene, which agreed to assist us with the research, development and clinical trials necessary for registration of RX-0201 in Asia. Under the agreement, we granted Rexgene an exclusive license, with right to sublicense, to make, have made, use, sell and import RX-0201 in Asia. In accordance with the agreement, Rexgene paid us a one-time fee of $1,500,000 in 2003.
On February 5, 2018, we entered into a royalty and release agreement with Next-BT, the successor in interest to Rexgene. In exchange for Next-BT terminating its rights to RX-0201 in Asia, we agreed to pay Next-BT a royalty in the low single digits of any net sales of RX-0201 we make in Asia and 50% of our licensing revenue related to licensing of RX-0201 in Asia, up to an aggregate of $5,000,000. The agreement will terminate upon the earlier of Next-BT’s receipt of $5,000,000 under the agreement, February 5, 2025 if Next-BT has received at least $3,000,000 under the agreement by that date, and the date after February 5, 2025 that Next-BT has received cumulative payments of $3,000,000 under the agreement.
Korea Research Institute of Chemical Technology (“KRICT”)
In June 2009, we entered into a license agreement with KRICT to acquire rights to all of KRICT’s intellectual property related to quinoxaline-piperazine derivatives, which includes RX-5902. We paid an initial license fee of $100,000 in July 2009, and will pay a one-time milestone payment of $1,000,000 to KRICT upon marketing approval from FDA for the first commercial product stemming from intellectual property (the “Milestone Payment”). Upon payment of the Milestone Payment all of the rights previously licensed to us will be transferred to us and the agreement will terminate. The agreement is terminable by either party for the other party’s material breach, subject to a 60-day cure period. To date, we have paid only the $100,000 initial license fee pursuant to this agreement.
The University of Maryland Baltimore (“UMB”)
In July 2013, we entered into an exclusive license agreement with UMB for a novel drug delivery platform, Nano-Polymer-Drug Conjugate Systems. This platform combines existing chemotherapeutic agents with a proprietary polymer carrier that contains a signaling moiety to direct the agents into a tumor. This agreement requires us to make payments to UMB if any other products developed from the licensed delivery platform achieve development milestones.
The Ohio State University
In October 2013, we entered into an exclusive license agreement with the Ohio State Innovation Foundation, an affiliate of The Ohio State University, for a novel oligonucleotide drug delivery platform, Lipid-Coated Albumin Nanoparticle (“LCAN”). The LCAN platform incorporates both cationic lipid and cationized albumin that can form an electrostatic complex with oligonucleotides and be co-encapsulated by lipids. The agreement requires us to make payments to the Ohio State Innovation Foundation if any products from the licensed delivery platform achieve development milestones.
Total Research and Development Costs
We have incurred research and development costs of $10,715,296, $10,089,149, and $12,148,226 for the years ended December 31, 2017, 2016 and 2015, respectively. Research and development costs primarily consist of clinical trials and pre-clinical development costs, as well as payroll costs for research and development personnel.
Employees
We currently have 17 full-time employees, all of whom are based either at our Rockville, Maryland office or our Gaithersburg, Maryland lab facility. Our employees are not covered by any collective bargaining agreement and we have never experienced a work stoppage. We believe our relationships with our employees are satisfactory.
Available Information
Under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), we are required to file annual, quarterly and current reports, proxy statements and other information with the SEC. Any document we file with the SEC may be read and copied at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at (800) SEC-0330 for further information about the public reference room. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
We make available, free of charge, on our website at www.rexahn.com our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K and all amendments thereto, as soon as reasonably practicable after they are filed with or furnished to the SEC. Investors are encouraged to access these reports and the other information about our business on our website. Information found on our website is not part of this Annual Report on Form 10-K (this “Annual Report”). We will also provide copies of this Annual Report, free of charge, upon written request to the Investor Relations Department at our main address, 15245 Shady Grove Road, Suite 455, Rockville MD, 20850.
Also posted on our website, and available in print upon written request of any shareholder to our Investor Relations Department, are the charters of the standing committees of our Board.
You should carefully consider the risks described below together with the other information included in this Form 10-K. Our business, financial condition or results of operations could be adversely affected by any of these risks. If any of these risks occur, the value of our common stock could decline.
Risks Related to Our Financial Position and Capital Needs
We currently have no product revenues, have incurred negative cash flows from operations since inception and will need to raise additional capital to operate our business.
To date, we have generated no product revenues and have incurred negative cash flow from operations. Until we receive approval from the FDA or other regulatory authorities for our drug candidates, we cannot sell our drugs and will not have product revenues. We expect to continue to incur significant development and other expenses related to our ongoing operations. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from the net proceeds of equity or debt offerings, cash on hand, licensing fees and grants, if any. If we are not able to raise sufficient funds, we will have to reduce our research and development activities. We will first reduce research and development activities associated with our pre-clinical compounds. To the extent necessary, we will then reduce our research and development activities related to some or all of our clinical stage product candidates.
Unforeseen events, difficulties, complications and delays may occur that could cause us to utilize our existing capital at a faster rate than projected, including the progress of our research and development efforts, the cost and timing of regulatory approvals and the costs of protecting our intellectual property rights. We may seek additional financing to implement and fund other drug candidate development, clinical trial and research and development efforts, including clinical trials for other new drug candidates, as well as other research and development projects.
We will need additional financing to continue to develop our drug candidates, which may not be available on favorable terms, if at all. If we are unable to secure additional financing in the future on acceptable terms, or at all, we may be unable to complete our planned pre-clinical and clinical trials or obtain approval of our drug candidates from the FDA and other regulatory authorities. In addition, we may be forced to reduce or discontinue product development or product licensing, reduce or forego sales and marketing efforts and forego attractive business opportunities in order to improve our liquidity to enable us to continue operations. Any additional sources of financing will likely involve the sale of our equity securities or securities convertible into our equity securities, which may have a dilutive effect on our stockholders.
We are not currently profitable and may never become profitable.
To date, we have generated no product revenues and have incurred negative cash flow from operations. Our accumulated deficit as of December 31, 2017 and 2016 was $140,318,712 and $115,024,209, respectively. For the years ended December 31, 2017, 2016 and 2015, we had net losses of $25,294,503, $9,307,345, and $14,384,556, respectively. Even if we succeed in developing and commercializing one or more of our drug candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future, based on the following considerations:
| · |
continued pre-clinical development and clinical trials for our current and new drug candidates;
|
| · |
finding and maintaining suitable partnerships to help us research, develop and commercialize drug candidates;
|
| · |
efforts to seek regulatory approvals for our drug candidates;
|
| · |
implementing additional internal systems and infrastructure;
|
| · |
in-licensing additional technologies to develop; and
|
| · |
hiring additional personnel or entering into relationships with third parties to perform functions that we are unable to perform on our own.
|
We also expect to continue to experience negative cash flow for the foreseeable future as we fund our operations and capital expenditures. Until we have the capacity to generate revenues, we are relying upon outside funding resources to fund our cash flow requirements. If these resources are depleted or unavailable, we may be unable to continue to expand our operations or otherwise capitalize on our business opportunities, and our business, financial condition and results of operations would be materially adversely affected.
We have a limited operating history, and we have not demonstrated an ability to commercialize drug candidates.
We are a clinical-stage company with a limited number of drug candidates. We currently do not have any products that have gained regulatory approval, and we have not demonstrated an ability to perform the functions necessary for the successful commercialization of any of our drug candidates. The successful commercialization of our drug candidates will require us to first perform a variety of functions, including:
| · |
conducting pre-clinical and clinical trials;
|
| · |
participating in regulatory approval processes;
|
| · |
formulating and manufacturing products; and
|
| · |
conducting sales and marketing activities.
|
To date, our operations have been limited to organizing and staffing the Company, acquiring, developing and securing our proprietary technology, and undertaking drug candidate research and development, including pre-clinical trials and clinical trials of our principal drug candidates. These operations provide a limited basis for assessing our ability to commercialize drug candidates.
If we fail to comply with the continued listing standards of NYSE American, our common stock could be delisted. If it is delisted, our common stock and the liquidity of our common stock would be impacted.
Our common stock is listed on NYSE American, and the continued listing of our common stock on NYSE American is subject to our compliance with a number of listing standards. For example, Section 1003(f)(v) of the NYSE American Company Guide provides that a company’s common stock may be delisted from NYSE American if it sells for a substantial period of time at a low price per share and the company fails to effect a reverse stock split or otherwise demonstrate sustained price improvement within a reasonable time after being notified that NYSE American deems such action to be appropriate under all the circumstances. Our common stock has previously traded at prices at which we expected that NYSE American could have delivered such a notice if the price did not increase. While our stock price thereafter increased, and while on May 5, 2017, we effected a one-for-ten reverse stock split of the outstanding shares of our common stock, there is no assurance that the market price of our common stock will remain at the level required to remain in compliance with NYSE American listing standards or that we will otherwise remain in compliance with NYSE American listing standards. Among other things, the NYSE American listing standards also have applicable provisions related to stockholders’ equity, disposal of assets and reduction of operations, and compliance with SEC and NYSE American regulations.
Delisting from NYSE American would adversely affect our ability to raise additional financing through the public or private sale of equity securities, significantly affect the ability of investors to trade our securities and negatively affect the value and liquidity of our common stock. Delisting also could have other negative results, including the potential loss of employee confidence, the loss of institutional investors or interest in business development opportunities. Moreover, we committed in connection with the sale of securities to use commercially reasonably efforts to maintain the listing of our common stock during such time that certain warrants are outstanding.
Risks Related to Our Business
Several of our drug candidates are in clinical trials, which are very expensive, time-consuming and difficult to design and implement.
Our drug candidates are in various stages of development and require extensive clinical testing. Such testing is expensive and time-consuming and requires specialized knowledge and expertise. RX-3117 entered a Phase 2a clinical trials in March 2016, September 2016, and November 2017 and RX-5902 entered a Phase 2a clinical trial in February 2017.
Human clinical trials are expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time-consuming, and the outcome is not certain; the results of pre-clinical studies and early clinical trials may not be predictive of the results of later-stage clinical trials. We estimate that clinical trials of our current drug candidates will take multiple years to complete. Furthermore, failure can occur at any stage of a clinical trial, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed or precluded by a number of factors, including:
| · |
delay or failure in reaching agreement with the FDA or a foreign regulatory authority on the design of a given trial, or in obtaining authorization to commence a trial;
|
| · |
delay or failure in reaching agreement on acceptable terms with prospective CROs and clinical trial sites;
|
| · |
delay or failure in obtaining approval of an IRB to conduct a clinical trial at a given site;
|
| · |
withdrawal of clinical trial sites from our clinical trials, including as a result of changing standards of care or the ineligibility of a site to participate;
|
| · |
delay or failure in recruiting and enrolling study subjects;
|
| · |
delay or failure in having subjects complete a clinical trial or return for post-treatment follow up;
|
| · |
clinical sites or investigators deviating from trial protocol, failing to conduct the trial in accordance with applicable regulatory requirements, or dropping out of a trial;
|
| · |
inability to identify and maintain a sufficient number of trial sites;
|
| · |
failure of third-party clinical trial managers to meet their contractual obligations or deadlines;
|
| · |
the need to modify a study protocol;
|
| · |
unforeseen safety issues;
|
| · |
emergence of dosing issues;
|
| · |
lack of effectiveness during clinical trials;
|
| · |
change in the standard of care of the indication being studied;
|
| · |
reliance on third-party suppliers for the clinical trial supply of drug candidates;
|
| · |
inability to monitor patients adequately during or after treatment;
|
| · |
lack of sufficient funding to finance the clinical trials; and
|
| · |
changes in governmental regulations or administrative action.
|
We, the FDA or an IRB may suspend a clinical trial at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our IND applications or the conduct of these trials. Additionally, we may have difficulty enrolling patients in our clinical trials. If we experience such difficulties, we may not be able to complete a clinical trial or we may experience significant delays in completing a clinical trial.
If the results of our clinical trials fail to support the approval of any of our drug candidates, the completion of development of that candidate may be significantly delayed, or we may be forced to abandon development altogether, which will significantly impair our ability to generate product revenues.
Even if our clinical trials are completed as planned, we cannot be certain that clinical results will support approval of our drug candidates. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing. The clinical trial process may fail to demonstrate that one or more of our drug candidates are safe and effective for indicated uses. As a result, we may have to conduct additional clinical trials or may decide to abandon a drug candidate, in which case we may never recognize any revenue related to such candidate. Standard of care treatments may change, which may require additional clinical trials. Repeating clinical trials or conducting additional clinical trials will increase our development costs and delay the filing of an NDA and, ultimately, delay our ability to commercialize our drug candidates and generate product revenues.
We may not obtain the necessary U.S. or worldwide regulatory approvals to commercialize our drug candidates, and we cannot guarantee how long it will take the FDA or other comparable regulatory agencies to review applications for our drug candidates.
We will need FDA approval to commercialize our drug candidates in the United States and approvals from the comparable regulatory authorities to commercialize our drug candidates in foreign jurisdictions.
The time it takes to obtain approval, either in the United States or foreign jurisdictions, is unpredictable, but typically takes many years, depending upon a variety of factors, including the type, complexity and novelty of the drug candidate. Obtaining approval requires substantial resources and is subject to regulatory authorities’ substantial discretion. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s development and may vary among jurisdictions. We cannot guarantee that any of our drug candidates will ultimately be approved by the FDA or any other regulatory authority, or the length of time obtaining approval will take. One of our drug candidates, RX-0201, is an ASO compound. To date, the FDA has approved relatively few ASO compounds. In addition, RX-0201 is in the drug class known as Akt-1 inhibitors that to date have not been approved by the FDA, and we have not submitted an NDA for any Akt-1 inhibitor.
Our product candidates could fail to receive regulatory approval from the FDA or a comparable foreign authority for a variety of reasons, including:
| · |
disagreement with the design or implementation of our clinical trials;
|
| · |
failure to demonstrate to the authority’s satisfaction that the product candidate is safe and effective for the proposed indication;
|
| · |
failure of clinical trial results to meet the level of statistical significance required for approval;
|
| · |
failure to demonstrate that the product’s benefits outweigh its risks;
|
| · |
disagreement with our interpretation of pre-clinical or clinical data; and
|
| · |
inadequacies in the manufacturing facilities or processes of third-party manufacturers.
|
The FDA or a comparable foreign authority may require us to conduct additional pre-clinical and clinical testing, which may delay or prevent approval and our commercialization plans or cause us to abandon the development program. Further, any approval we receive may be for fewer or more limited indications than we request, may not include labeling claims necessary for successful commercialization of the product candidate, or may be contingent upon our conducting costly post-marketing clinical trials. Any of these scenarios could materially harm the commercial prospects of a product candidate.
Even if our product candidates obtain approval, they may face future development and regulatory difficulties that can negatively affect commercial prospects.
Even if we obtain approval for a product candidate, it would be subject to ongoing regulatory requirements and restrictions of the FDA and comparable regulatory authorities regarding manufacturing, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting. Failure by us or any of the third parties on which we rely to meet those requirements can lead to enforcement action, among other consequences, that could significantly impair our ability to successfully commercialize a given product. If the FDA or a comparable regulatory authority becomes aware of new safety information, it can impose additional restrictions on how the product is marketed or may seek to withdraw marketing approval altogether.
There is no assurance that any of our product candidates that has received or will receive orphan drug designation will subsequently obtain orphan drug exclusivity, or that any such exclusivity will provide the desired benefit.
Although we have obtained orphan drug designation for several uses of RX-0201 and one use of RX-3117 and in the future may obtain additional orphan drug designation for these or other product candidates, we are not assured of being awarded orphan drug exclusivity or realizing the benefits of such exclusivity, even if any of these products is approved for its orphan-designated use. If another company also holding orphan drug designation for a product containing the same active moiety intended for the same rare disease or condition receives approval before our orphan-designated product, approval of our product could be precluded for seven years because of that product’s orphan drug exclusivity, unless we could demonstrate our product to be clinically superior to the earlier-approved product. Similarly, even if our orphan designated drug were approved first and awarded seven-year orphan drug exclusivity, it would not block approval of the other product if that product were shown to be clinically superior, or if we fail to assure a sufficient quantity of our orphan drug. Additionally, because orphan drug exclusivity is product- and indication-specific, it does not prevent approval of another drug for the same orphan indication or the same drug for a different use.
If physicians and patients do not accept and use our drugs, our ability to generate revenue from sales of our products will be materially impaired.
Even if the FDA approves our drug candidates, physicians and patients may not accept and use them. Future acceptance and use of our products will depend upon a number of factors including, but not limited to:
| · |
awareness of a drug’s availability and benefits;
|
| · |
perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drugs;
|
| · |
pharmacological benefit and cost-effectiveness of our products relative to competing products;
|
| · |
availability of reimbursement for our products from government or other third-party payors;
|
| · |
effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any; and
|
| · |
the price at which we sell our products.
|
Because we expect sales of our current drug candidates, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
Even if we are able to commercialize any of our product candidates, these products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates even if our product candidates obtain marketing approval.
Our ability to commercialize any products successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available in a timely manner from government third-party payors, including governmental healthcare programs such as Medicare and Medicaid, commercial health insurers and managed care organizations. Government authorities and other third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. Third-party payors may limit coverage to specific products on an approved list, or formulary, which may not include all of the FDA-approved products for a particular indication. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government healthcare programs and other third-party payors are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy, and have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available promptly or at all for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases. Limited coverage may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any product candidate for which we obtain marketing approval.
Payors also are increasingly considering new metrics as the basis for reimbursement rates, such as ASP, AMP and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS surveys and publishes retail community pharmacy acquisition cost information in the form of NADAC files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. It may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payors to cover our products for which we receive regulatory approval.
If we successfully commercialize any of our products, we may participate in the Medicaid Drug Rebate program. Participation is required for federal funds to be available for our products under Medicaid and Medicare Part B. Under the Medicaid Drug Rebate Program, we would be required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Part B of the Medicare program.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients.
In addition, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, a manufacturer also must participate in the FSS pricing program, established by Section 603 of the VHCA. Under this program, the manufacturer is obligated to make its covered drugs available for procurement on an FSS contract and charge a price to the Big Four agencies that is no higher than the statutory Federal Ceiling Price. Moreover, pursuant to DHA regulations, manufacturers must provide rebates on utilization of their covered drugs that are dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. The formula for determining the rebate is established in the regulations and is based on the difference between the annual Non-FAMP and the FCP in effect on the dispense date (these price points are required to be calculated by us under the VHCA). The requirements under the 340B, FSS, and TRICARE programs could reduce the revenue we may generate from any products that are commercialized in the future and could adversely affect our business and operating results.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Limited coverage may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. Third-party payors also may seek additional clinical evidence, including expensive pharmacoeconomic studies beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations, before covering our products for those patients. If reimbursement is available only for limited indications, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Changes in healthcare law and implementing regulations, including those based on recently enacted and future legislation, as well as changes in healthcare policy, may increase the difficulty and cost for us to commercialize our product candidates and affect the prices we may obtain.
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidate for which we obtain marketing approval. The United States government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
In recent years, Congress has considered reductions in Medicare reimbursement levels for drugs administered by physicians. CMS also has authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products, which in turn would affect the price we can receive for those products. While Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “Affordable Care Act”). This law substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum Medicaid rebate for both branded and generic drugs, expanded the 340B program, and revised the definition of AMP. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service Medicaid utilization, to include the utilization of Medicaid managed care organizations as well and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the amount of rebates due on those drugs. On February 1, 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016.
Moreover, certain legislative changes to and regulatory changes under the Affordable Care Act have occurred in the 115th United States Congress and under the Trump Administration. For example, the Tax Cuts and Jobs Act, enacted on December 22, 2017, eliminated the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code of 1986, commonly referred to as the individual mandate, beginning in 2019. Additional legislative changes to and regulatory changes under the Affordable Care Act remain possible. Any such changes could decrease the number of individuals with health coverage. We expect that the Affordable Care Act, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to successfully commercialize our product candidates, if approved.
The Affordable Care Act requires pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government. Each individual pharmaceutical manufacturer pays a prorated share of the branded prescription drug fee of $4.1 billion in 2018, based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. Furthermore, the new law requires manufacturers to provide a 50% discount off the negotiated price of prescriptions filled by beneficiaries in the Medicare Part D coverage gap, referred to as the “donut hole.”
The Affordable Care Act also expanded the Public Health Service’s 340B drug pricing discount program. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. The Affordable Care Act expanded the 340B program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by the Affordable Care Act. The Affordable Care Act exempts “orphan drugs”—those designated under section 526 of the Food, Drug, and Cosmetic Act—from the ceiling price requirements for these newly-eligible entities. Because the 340B ceiling price is determined based on AMP and Medicaid drug rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discounts to increase.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. For example, recent legislative enactments have resulted in Medicare payments being subject to a two percent reduction, referred to as sequestration, until 2027. Continuation of sequestration or enactment of other reductions in Medicare reimbursement for drugs could affect our ability to achieve a profit on any candidate products that are approved for marketing.
We expect that the Affordable Care Act, as well as other healthcare reform measures that have and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product and could seriously harm our future revenues. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
If we are able to successfully commercialize any of our products and if we participate in the Medicaid drug rebate program or other governmental pricing programs, failure to comply with reporting and payment obligations under these programs could result in additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
The Medicaid Drug Rebate Program and other governmental pricing programs require participating manufacturers to report pricing data to the government. Pricing calculations vary among products and programs and include average manufacturer price and best price for the Medicaid Drug Rebate Program, average sales price for certain categories of drugs that are paid under Part B of the Medicare program, and FCP and non-FAMP for the FSS pricing program. If we successfully commercialize any of our products and participate in such governmental pricing programs, we will be liable for errors associated with our submission of pricing data. That liability could be significant. For example, if we are found to have knowingly submitted false average manufacturer price, average sales price, best price, or Non-FAMP information to the government, we may be liable for civil monetary penalties in the amount of $181,071 per item of false information. If we are found to have made a misrepresentation in the reporting of average sales price, the statute provides for civil monetary penalties of up to $13,066 for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit monthly/quarterly average manufacturer price, average sales price, and best price, and quarterly/annual Non-FAMP data on a timely basis could result in a civil monetary penalty of $18,107 per day for each day the information is late beyond the due date. Such failure also could be grounds for other sanctions, such as termination from the Medicaid Drug Rebate Program.
If we fail to comply with data protection laws and regulations, we could be subject to government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity, which could negatively affect our operating results and business.
We are subject to data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the U.S., numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), govern the collection, use, disclosure, and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions and create liability for us (which could include civil and/or criminal penalties), private litigation and/or adverse publicity that could negatively affect our operating results and business. In addition, we may obtain health information from third parties (e.g., healthcare providers who prescribe our products) that are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA—other than potentially with respect to providing certain employee benefits—we could be subject to criminal penalties if we knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. HIPAA generally requires that healthcare providers and other covered entities obtain written authorizations from patients prior to disclosing protected health information of the patient (unless an exception to the authorization requirement applies). If authorization is required and the patient fails to execute an authorization or the authorization fails to contain all required provisions, then we may not be allowed access to and use of the patient’s information and our research efforts could be impaired or delayed. Furthermore, use of protected health information that is provided to us pursuant to a valid patient authorization is subject to the limits set forth in the authorization (e.g., for use in research and in submissions to regulatory authorities for product approvals). In addition, HIPAA does not replace federal, state, international or other laws that may grant individuals even greater privacy protections.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse, transparency and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
| · |
the federal Anti-Kickback Law prohibits persons from, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, the referral of an individual for the furnishing or arranging for the furnishing, or the purchase, lease or order, or arranging for or recommending purchase, lease or order, any good or service for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
|
| · |
the federal civil False Claims Act imposes penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government;
|
| · |
HIPAA imposes criminal liability for knowingly and willfully executing a scheme to defraud any healthcare benefit program, knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a health care offense, or knowingly and willfully making false statements relating to healthcare matters;
|
| · |
HIPAA and its implementing regulations also impose obligations on certain covered entity health care providers, health plans and health care clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
|
| · |
the federal Open Payments program, created under Section 6002 of the Affordable Care Act and its implementing regulations, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the CMS information related to “payments or other transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually CMS ownership and investment interests held by physicians (as defined above) and their immediate family members; and
|
| · |
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to certain healthcare providers; state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws that govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
|
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, that person or entity may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. For a fuller discussion of the applicable anti-kickback fraud and abuse, transparency and other healthcare laws and regulations applicable to our business, see Item 1, “Description of Business – Government Regulation.”
Developments by competitors may render our products or technologies obsolete or non-competitive.
We compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies as well as academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as more experience in:
| · |
developing drugs;
|
| · |
undertaking pre-clinical testing and human clinical trials;
|
| · |
obtaining FDA and other regulatory approvals of drugs;
|
| · |
formulating and manufacturing drugs; and
|
| · |
launching, marketing and selling drugs.
|
Large pharmaceutical companies currently sell both generic and proprietary compounds for the treatment of cancer. In addition, companies developing oncology therapies represent substantial competition. Many of these organizations have substantially greater capital resources, larger research and development staff and facilities, history in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to attract qualified personnel, parties for acquisitions, joint ventures or other collaborations. Our competitors may succeed in obtaining regulatory approval of their products more rapidly than we are able to, obtaining patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates, or developing products that are more effective and/or safer than ours, any of which could render our product candidates less competitive prior to recovery by us of expenses incurred with respect to their development and could lead us to alter our business plans or development strategies. For example, in response to the changing treatment landscape for renal cell carcinoma (“RCC”) patients over the past two years with the approval of new therapies by the FDA, in February 2018, we announced plans to discontinue the internally funded programs of RX-0201 and ceased enrolling patients in a Phase 2a proof-of-concept clinical trial of RX-0201 in patients with metastatic RCC.
If we are unable to successfully manage our growth, our business may be harmed.
In addition to our own internally developed drug candidates, we are actively seeking opportunities to in-license compounds in oncology and other therapeutic areas that are strategic additions to our product pipeline. Such additional drug candidates could significantly increase our capital requirements and place further strain on our resources, including on the time of our existing personnel, which may delay or otherwise adversely affect the development of our existing drug candidates. As of December 31, 2017, we had 17 full-time employees. We may need to hire more employees as our product pipeline and operations expand, further increasing the size of our organization and related expenses. If we are unable to manage our growth effectively, we may not efficiently use our resources, which may delay the development of our drug candidates and negatively impact our business, results of operations and financial condition.
We may not be able to attract and retain qualified personnel necessary for the development and commercialization of our drug candidates. Our success may be negatively impacted if key personnel leave.
Attracting and retaining qualified personnel is critical to our future success. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot assure you that we will be successful in engaging personnel with the skills and experience to support our business and research and development activities.
Our key personnel, especially Dr. Peter Suzdak, our Chief Executive Officer, Mr. Douglas Swirsky, our President and Chief Financial Officer, Dr. Ely Benaim, our Chief Medical Officer, and Dr. Lisa Nolan, our Chief Business Officer, provide critical technical knowledge and expertise. The loss of, Dr. Suzdak, Mr. Swirsky, Dr. Benaim, Dr. Nolan or any of the other members of our management team could result in delays in product development and diversion of management resources, which could adversely affect our operating results. We do not have “key person” life insurance policies for any of our executive officers.
The recently passed comprehensive tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law new legislation that significantly revises the Internal Revenue Code of 1986, as amended. The newly enacted federal income tax law, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, and limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain, and our business and financial condition could be adversely affected. The impact of this tax reform on holders of our common stock is also uncertain and could be adverse.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of medical products entail an inherent risk of product liability. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. Although we currently carry clinical trial insurance and product liability insurance we, or any collaborators, may not be able to maintain such insurance at a reasonable cost. Even if our agreements with any future collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claims arise.
Risks Related to Reliance on Third Parties
Much of our drug development program depends upon third parties, and thus the conduct and completion of our clinical trials are, to some extent, beyond our control.
We have engaged third-party CROs and other investigators and collaborators, such as universities, medical institutions and other life science companies, to conduct our pre-clinical studies, toxicology studies and clinical trials, and to pursue development for our product candidates. For example, in February 2018, we entered into a research collaboration and license agreement with Zhejiang Haichang Biotechnology Co., Ltd. (“Haichang”) pursuant to which Haichang will develop a nano-liposomal formulation of RX-0201 and will conduct certain pre-clinical and clinical activities through completion of a Phase 2a proof-of-concept clinical trial in hepatic cell carcinoma in China. Engaging third parties, or collaborating with third parties, is typical practice in our industry. However, relying on such organizations means that the conduct of clinical trials and other studies, and the completion of these trials and studies, is not within our direct control. Trials and studies may be delayed due to circumstances outside our control, and such delays may result in additional expenses for us.
While we make efforts to oversee the work of third-party contractors, these collaborators are not our employees, and we cannot control the effort, time or other resources that they devote to our programs. Third parties may not assign priority to our programs or pursue them as diligently as we would if we were undertaking them ourselves.
If outside collaborators fail to devote sufficient time and resources to our drug-development programs, or if their performance is substandard, the approval of our FDA applications and introduction of new drugs to the market may be delayed or unsuccessful. For example, the success of the Haichang agreement depends on, among other things, the skills, experience and efforts of Haichang, Haichang’s commitment to the arrangement, and the financial condition of Haichang, all of which are beyond our control. In the event that Haichang fails to successfully develop or commercialize RX-0201, including due to early termination of the Haichang agreement, our ability to obtain license fees, milestone payments and royalties would be adversely affected, which could have an adverse effect on our financial condition and results of operation. Our collaborators may also have relationships with other commercial entities, some of which may compete with us. If our collaborators assist our competitors at our expense, our competitive position would be harmed.
We rely exclusively on third parties to formulate and manufacture our drug candidates, which exposes us to a number of risks that may delay development, regulatory approval and commercialization of our products or result in higher product costs.
We have no experience in drug formulation or manufacturing and we lack the resources and expertise to formulate or manufacture our own drug candidates internally. Therefore, we rely on third-party expertise to support us in this area. We have entered into contracts with third-party manufacturers to manufacture, supply, store and distribute supplies of our drug candidates for our clinical trials. If any of our drug candidates receives FDA approval, we expect to rely on third-party contractors to manufacture our drugs. We have no current plans to build internal manufacturing capacity for any product candidate, and we have no long-term supply arrangements.
Our reliance on third-party manufacturers exposes us to potential risks, such as the following:
| · |
We may be unable to contract with third-party manufacturers on acceptable terms, or at all, because the number of potential manufacturers is limited. Potential manufacturers of any product candidate that is approved will be subject to FDA compliance inspections and any new manufacturer would have to be qualified to produce our products;
|
| · |
Our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical and commercial needs, if any;
|
| · |
Our third-party manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials through completion or to successfully produce, store and distribute our commercial products, if approved;
|
| · |
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA and other government agencies to ensure compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards, but we may ultimately be responsible for any of their failures;
|
| · |
If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to such improvements; and
|
| · |
A third-party manufacturer may gain knowledge from working with us that could be used to supply one of our competitors with a product that competes with ours.
|
Each of these risks could delay or have other adverse impacts on our clinical trials and the approval and commercialization of our drug candidates, potentially resulting in higher costs, reduced revenues or both.
We have no experience selling, marketing or distributing products and currently no internal capability to do so.
We currently have no sales, marketing or distribution capabilities. While we intend to have a role in the commercialization of our products, we do not anticipate having the resources in the foreseeable future to develop global sales and marketing capabilities for all of our proposed products. Our future success depends, in part, on our ability to enter into and maintain collaborative relationships with other companies that have sales, marketing and distribution capabilities, a strategic interest in the products under development, and the ability to successfully market and sell our products. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with the necessary expertise. We cannot assure you that we will be able to establish or maintain relationships with third-party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, as well as the terms of our agreements with such third parties, which cannot be predicted at this early stage of our development. We cannot assure you that such efforts will be successful. In addition, we cannot assure you that we will be able to market and sell our products in the United States or overseas.
Risks Related to Our Intellectual Property
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish, and our business and competitive position would suffer.
Our success, competitive position and future revenues will depend in part on our ability and the abilities of our licensors and licensees to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We have an active patent protection program that includes filing patent applications on new compounds, formulations, delivery systems and methods of making and using products and prosecuting these patent applications in the United States and abroad. As patents issue, we also file continuation applications as appropriate. Although we have taken steps to build a strong patent portfolio, we cannot predict:
| · |
the degree and range of protection any patents will afford us against competitors, including whether third parties find ways to invalidate or otherwise circumvent our licensed patents;
|
| · |
if and when patents will issue in the United States or any other country;
|
| · |
whether or not others will obtain patents claiming aspects similar to those covered by our licensed patents and patent applications;
|
| · |
whether we will need to initiate litigation or administrative proceedings to protect our intellectual property rights, which may be costly whether we win or lose;
|
| · |
whether any of our patents will be challenged by our competitors alleging invalidity or unenforceability and, if opposed or litigated, the outcome of any administrative or court action as to patent validity, enforceability or scope;
|
| · |
whether a competitor will develop a similar compound that is outside the scope of protection afforded by a patent or whether the patent scope is inherent in the claims modified due to interpretation of claim scope by a court;
|
| · |
whether there were activities previously undertaken by a licensor that could limit the scope, validity or enforceability of licensed patents and intellectual property; or
|
| · |
whether a competitor will assert infringement of its patents or intellectual property, whether or not meritorious, and what the outcome of any related litigation or challenge may be.
|
Our success also depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our licensors, sublicensees and contractors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, we require all employees to enter into agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired, and our business and competitive position would suffer.
Due to legal and factual uncertainties regarding the scope and protection afforded by patents and other proprietary rights, we may not have meaningful protection from competition.
Our long-term success will substantially depend upon our ability to protect our proprietary technologies from infringement, misappropriation, discovery and duplication and avoid infringing the proprietary rights of others. Our patent rights, and the patent rights of biopharmaceutical companies in general, are highly uncertain and include complex legal and factual issues. These uncertainties also mean that any patents that we own or may obtain in the future could be subject to challenge, and even if not challenged, may not provide us with meaningful protection from competition. Patents already issued to us or our pending applications may become subject to dispute, and any dispute could be resolved against us.
In connection with the process of seeking patent protection for RX-5902 in Japan, we filed a patent application including claims covering RX-5902 with the Japanese Patent Office (“JPO”) for examination. The JPO initially agreed that the claims covering the compound for RX-5902 were allowable, but as a result of a mistake in the patent application filing as prepared and submitted by our Japanese patent attorney and incomplete review by the JPO’s patent examiner, the JPO issued a decision to grant a patent with claims that did not include RX-5902’s chemical structure. We appealed this decision with the JPO and requested withdrawal of the ‘decision to grant’ so that the correct claims would be allowed, but the JPO refused to withdraw its decision. As a result, and in accordance with Japanese law and procedure for appealing patent application decisions, we have filed a lawsuit against the JPO in Tokyo District Court to cause the JPO to reverse its decision to grant the errant patent and to allow a patent that includes claims covering RX-5902. The patent application at issue remains pending subject to the outcome of this action. However, there can be no guarantee that we will be successful in winning the appeal to correct the error in the patent registration that would exclude the compound for RX-5902. While the composition of matter patent on RX-5902’s structure remains pending in Japan, we have also filed, or intend to file, additional patents covering method of use and manufacturing process that would extend to 2035/2036 if approved. We also expect that RX-5902 will be covered by regulatory exclusivity up to ten years post approval.
If we infringe the rights of third parties, we could be prevented from selling products and be forced to defend against litigation and pay damages.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and may have to:
| · |
obtain licenses, which may not be available on commercially reasonable terms, if at all;
|
| · |
redesign our products or processes to avoid infringement;
|
| · |
stop using the subject matter claimed in patents held by others, which could cause us to lose the use of one or more of our drug candidates;
|
| · |
pay damages; or
|
| · |
defend litigation or administrative proceedings that may be costly whether we win or lose and that could result in a substantial diversion of our management resources.
|
Although we have not received any claims of infringement by any third parties to date, we expect that as our drug candidates move further into clinical trials and commercialization and our public profile is raised, we may be subject to such claims.
Risks Related to Ownership of Our Common Stock
An investment in shares of our common stock is very speculative and involves a very high degree of risk.
To date, we have generated no revenues from product sales and only minimal revenues from a research agreement with a minority stockholder and interest on bank account balances and short-term investments. Our accumulated deficit as of December 31, 2017 and 2016 was $140,318,712 and $115,024,209 respectively. For the years ended December 31, 2017, 2016 and 2015, we had net losses of $25,294,503, $9,307,345 and $14,384,556 respectively, partially as a result of expenses incurred through a combination of research and development activities related to the various technologies under our control and expenses supporting those activities. Until we receive approval from the FDA and other regulatory authorities for our drug candidates, we cannot sell our drugs and will not have product revenues.
The market price of our common stock may fluctuate significantly.
The market price of our common stock may fluctuate significantly in response to factors, some of which are beyond our control, such as:
| · |
the announcement of new products or product enhancements by us or our competitors;
|
| · |
changes in our relationships with our licensors or other strategic partners;
|
| · |
developments concerning intellectual property rights and regulatory approvals;
|
| · |
variations in our and our competitors’ results of operations;
|
| · |
changes in earnings estimates or recommendations by securities analysts;
|
| · |
changes in the structure of healthcare payment systems; and
|
| · |
developments and market conditions in the pharmaceutical and biotechnology industries.
|
Further, the stock market, in general, and the market for biotechnology companies, in particular, have experienced extreme price and volume fluctuations. Continued market fluctuations could result in extreme volatility in the price of our common stock, which may be unrelated or disproportionate to our operating performance and which could cause a decline in the value of our common stock. You should also be aware that price volatility might be worse if the trading volume of our common stock is low.
We will require additional capital funding the receipt of which may impair the value of our common stock.
Our future capital requirements depend on many factors, including our research, development, sales and marketing activities. We will need to raise additional capital through public or private equity or debt offerings or through arrangements with strategic partners or other sources in order to continue to develop our drug candidates. There can be no assurance that additional capital will be available when needed or on terms satisfactory to us, if at all. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution and the new equity securities may have greater rights, preferences or privileges than our existing common stock.
We have not paid dividends to our stockholders in the past, and we do not anticipate paying dividends to our stockholders in the foreseeable future.
We have not declared or paid cash dividends on our common stock. We currently intend to retain all future earnings, if any, to fund the continuing operation of our business, and therefore we do not anticipate paying dividends on our common stock in the foreseeable future. As a result, you will not realize any income from an investment in our common stock until and unless you sell your shares at a profit.
We may be subject to securities litigation, which is expensive and could divert management attention.
The market price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and direct our management’s attention from other business concerns, which could seriously harm our business.
None
We lease approximately 7,193 square feet of office space in Rockville, Maryland. We also lease approximately 1,100 square feet of laboratory space in Gaithersburg, Maryland. The laboratory space is equipped with the requisite laboratory services required to conduct our business and we believe our existing facilities are adequate to meet our needs for the foreseeable future. The office lease, which commenced on June 29, 2009, expires in June 2019. The laboratory lease, which commenced on July 1, 2015, expires in June 2020. We do not own any real property.
None
Not Applicable
PART II
|
Market for Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
|
Our common stock is traded on NYSE American, under the ticker symbol “RNN”. As of March 9, 2018, there were approximately 54 stockholders of record of our common stock. The following table sets forth the high and low sales prices of our common shares as reported on NYSE American during the periods indicated.
|
Period
|
High ($)
|
Low ($)
|
||||||
|
2016
|
||||||||
|
First Quarter
|
4.25
|
2.60
|
||||||
|
Second Quarter
|
3.42
|
2.40
|
||||||
|
Third Quarter
|
2.80
|
2.02
|
||||||
|
Fourth Quarter
|
2.29
|
1.27
|
||||||
|
2017
|
||||||||
|
First Quarter
|
5.62
|
1.36
|
||||||
|
Second Quarter
|
7.10
|
2.80
|
||||||
|
Third Quarter
|
3.06
|
1.70
|
||||||
|
Fourth Quarter
|
3.19
|
1.69
|
||||||
Reverse Stock Split
On May 5, 2017, we effected a one-for-ten reverse stock split of the outstanding shares of our common stock, together with a corresponding proportional reduction in the number of authorized shares of our capital stock. All share information contained in this report, including the sales prices listed above, has been retroactively adjusted to reflect the effects of the reverse split.
Dividends
We have not paid any cash dividends on common stock and do not expect to do so in the foreseeable future. We anticipate that any earnings generated from future operations will be used to finance our operations. No restrictions exist upon our ability to pay dividends.
Purchase of Equity Securities by the Issuer and Affiliated Purchasers
There were no repurchases of equity securities in 2017.
Recent Sales of Unregistered Equity Securities
None
Performance Graph
The following graph compares the cumulative total stockholder return on $100 of our common stock for the period beginning December 31, 2012 through December 31, 2017 with the cumulative total return over such period for an identical investment in (i) the NYSE Arca Biotechnology Index or (ii) the NYSE American Composite Index. This graph and accompanying text is not deemed to be “filed” with the SEC or subject to the liabilities of Section 18 of the Exchange Act, and the graph shall not be deemed to be incorporated by reference into any prior or subsequent filing by us under the Securities Act or the Exchange Act.
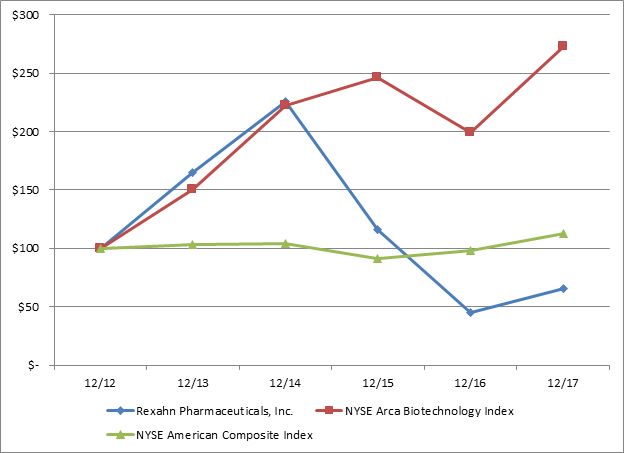
The following selected data should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements included elsewhere in this Annual Report.
|
|
For the Year Ended December 31,
|
|||||||||||||||||||
|
Statement of Operations Data:
|
2017
|
2016
|
2015
|
2014
|
2013
|
|||||||||||||||
|
Revenues
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||||||
|
Expenses:
|
||||||||||||||||||||
|
General and administrative
|
6,639,421
|
6,324,236
|
6,115,210
|
6,253,328
|
4,725,699
|
|||||||||||||||
|
Research and development
|
10,715,296
|
10,089,149
|
12,148,226
|
7,015,901
|
3,253,139
|
|||||||||||||||
|
Total expenses
|
17,354,717
|
16,413,385
|
18,263,436
|
13,269,229
|
7,978,838
|
|||||||||||||||
|
Loss from operations
|
(17,354,717
|
)
|
(16,413,385
|
)
|
(18,263,436
|
)
|
(13,269,229
|
)
|
(7,978,838
|
)
|
||||||||||
|
Other Income (Expense), net
|
(7,939,786
|
)
|
7,106,040
|
3,878,880
|
(5,252,372
|
)
|
(1,520,586
|
)
|
||||||||||||
|
Net Loss
|
$
|
(25,294,503
|
)
|
$
|
(9,307,345
|
)
|
$
|
(14,384,556
|
)
|
$
|
(18,521,601
|
)
|
$
|
(9,499,424
|
)
|
|||||
|
Net Loss per share, basic and diluted
|
$
|
(0.92
|
)
|
$
|
(0.43
|
)
|
$
|
(0.79
|
)
|
$
|
(1.05
|
)
|
$
|
(0.74
|
)
|
|||||
|
Weighted average shares outstanding, basic and diluted
|
27,390,527
|
21,744,740
|
18,238,822
|
17,610,697
|
12,864,929
|
|||||||||||||||
|
|
||||||||||||||||||||
|
|
As of December 31,
|
|||||||||||||||||||
|
Balance Sheet Data:
|
2017
|
2016
|
2015
|
2014
|
2013
|
|||||||||||||||
|
Cash, Cash Equivalents, and Marketable Securities
|
$
|
26,831,095
|
$
|
20,315,580
|
$
|
23,439,526
|
$
|
32,698,296
|
$
|
18,788,031
|
||||||||||
|
Working Capital(1)
|
$
|
24,901,710
|
$
|
19,041,597
|
$
|
22,000,046
|
$
|
30,970,020
|
$
|
18,361,438
|
||||||||||
|
Total Assets
|
$
|
28,287,881
|
$
|
21,043,532
|
$
|
24,805,029
|
$
|
33,533,060
|
$
|
19,556,498
|
||||||||||
|
Warrant Liabilities
|
$
|
7,853,635
|
$
|
1,573,366
|
$
|
2,739,163
|
$
|
3,768,351
|
$
|
5,034,058
|
||||||||||
|
Accumulated Deficit
|
$
|
(140,318,712
|
)
|
$
|
(115,024,209
|
)
|
$
|
(105,716,864
|
)
|
$
|
(91,332,308
|
)
|
$
|
(72,810,707
|
)
|
|||||
|
Total Stockholders' Equity
|
$
|
16,768,596
|
$
|
17,058,462
|
$
|
18,775,548
|
$
|
26,580,491
|
$
|
12,625,488
|
||||||||||
|
Common shares outstanding
|
31,725,114
|
23,736,878
|
19,741,378
|
17,825,331
|
14,671,772
|
|||||||||||||||
| (1) |
Working Capital defined as current assets less current liabilities
|
You should read the following discussion and analysis of our results of operations, financial condition and liquidity in conjunction with our financial statements and the related notes, which are included in this Annual Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report, including information with respect to our plans and strategies for our business, statements regarding the industry outlook, our expectations regarding the future performance of our business, and the other non-historical statements contained herein are forward-looking statements. See “Cautionary Statement Regarding Forward‑Looking Statements.” You should also review the “Risk Factors” section under this Item 1A of this Annual Report for a discussion of important factors that could cause actual results to differ materially from the results described herein or implied by such forward-looking statements.
OVERVIEW
We are a clinical stage biopharmaceutical company dedicated to the discovery, development and commercialization of innovative treatments for cancer. Our mission is to improve the lives of cancer patients by developing next-generation cancer therapies that are designed to maximize efficacy and minimize the toxicity and side effects traditionally associated with cancer treatment. Our clinical pipeline features two product candidates in Phase 2 clinical development and additional compounds in pre-clinical development. Our strategy is to continue building a significant pipeline of innovative oncology product candidates that we will commercialize alone or with partners.
Since our inception, our efforts and resources have been focused primarily on developing our pharmaceutical technologies, raising capital and recruiting personnel. We have no product sales to date, and we will not generate any product sales until we receive approval from the FDA or equivalent foreign regulatory bodies to begin selling our pharmaceutical candidates. Our major sources of working capital have been proceeds from various private and public financings, and licensing and collaboration agreements with our strategic investors and partners.
On May 5, 2017 we effected a one-for-ten reverse stock split of the outstanding shares of our common stock, together with a corresponding proportional reduction in the number of authorized shares of our capital stock. See Note 10, “Common Stock—Reverse Stock Split,” in the Notes to the Financial Statements of this Annual Report.
Critical Accounting Policies
A “critical accounting policy” is one which is both important to the portrayal of our financial condition and results and requires our management’s most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Our accounting policies are in accordance with U.S. generally accepted accounting principles and their basis of application is consistent with that of the previous year. Our significant estimates include assumptions made in estimating the fair values of stock-based compensation, warrant liabilities, marketable securities, and our assessment relating to costs incurred on research and development contracts.
Research and Development
Research and development costs are expensed as incurred. Research and development expenses consist primarily of third party service costs under research and development agreements, salaries and related personnel costs, as well as stock-based compensation related to these costs, costs to acquire pharmaceutical products and product rights for development and amounts paid to CROs, hospitals and laboratories for the provision of services and materials for drug development and clinical trials.
Costs incurred in obtaining the license rights to technology in the research and development stage that have no alternative future uses and are for unapproved product compounds are expensed as incurred.
We are required to estimate our accrued expenses. This process involves reviewing open contracts and purchase orders, communicating with our personnel to identify services performed on our behalf and estimating the level of service performed and the associated cost incurred when we have not yet been invoiced or otherwise notified of the actual cost. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. We estimate our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. Examples of estimated accrued research and development expenses include fees paid to:
| · |
CROs and investigative sites in connection with clinical studies;
|
| · |
vendors in connection with product manufacturing, development, and distribution of clinical supplies; and
|
| · |
vendors in connection with preclinical development activities.
|
We record expenses related to clinical studies and manufacturing development activities based on our estimates of the services received and efforts expended pursuant to contracts with multiple CROs and manufacturing vendors. The financial terms of these agreements are subject to negotiation, vary from contract to contract, and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the expense. Payments under some of these contracts depend on factors such as the successful enrollment of subjects and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed, enrollment of subjects, number of sites activated and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrued or prepaid expense balance accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, if our estimates of the status and timing of services performed differ from the actual status and timing of services performed, we may report amounts that are too high or too low in any particular period. To date, there have been no material differences from our estimates to the amounts actually incurred.
Fair Value of Financial Instruments
The carrying amounts reported in the accompanying financial statements for cash and cash equivalents, and accounts payable and accrued expenses approximate fair value because of the short‑term maturity of these financial instruments. The fair value methodology for our warrant liabilities and marketable securities is described in detail in Item 8 of this Annual Report.
Income Taxes
We account for income taxes in accordance with Accounting Standards Codification (“ASC”) 740, “Income Taxes.” Deferred tax assets and liabilities are recorded for differences between the financial statement and tax basis of the assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates. ASC 740 requires that a valuation allowance be established when it is more likely than not that all portions of a deferred tax asset will not be realized. A review of all positive and negative evidence needs to be considered, including a company’s current and past performance, the market environment in which the company operates, length of carryback and carryforward periods and existing contracts that will result in future profits. Income tax expense is recorded for the amount of income tax payable or refundable for the period, increased or decreased by the change in deferred tax assets and liabilities during the period.
As a result of our significant cumulative losses, we determined that it was appropriate to establish a valuation allowance for the full amount of our net deferred tax assets.
The calculation of our tax liabilities involves the inherent uncertainty associated with the application of complex tax laws. We are subject to examination by various taxing authorities. We believe that as a result of our losses sustained to date, any examination would result in a reduction of our net operating loss carryforward rather than a tax liability. As such, we have not provided for additional taxes estimated under ASC 740.
Warrant Liabilities
We record warrant liabilities at fair value due to provisions in our warrant agreements, as discussed further in Note 12, Warrants, in the Notes to the Financial Statements in this Annual Report. We reevaluate the fair value of our warrants at each reporting period, and changes in the fair value between reporting periods is recorded as “unrealized gain (loss) on fair value of warrants” in the statement of operations.
Stock-Based Compensation
In accordance with ASC 718, “Stock Compensation,” compensation costs related to share-based payment transactions, including employee stock options, are to be recognized in the financial statements. In addition, we adhere to the guidance set forth within SEC Staff Accounting Bulletin No. 107 (“SAB 107”), which provides the Staff’s views regarding the interaction between ASC 718 and certain SEC rules and regulations, and provides interpretations with respect to the valuation of share-based payments for public companies.
We estimate the fair value of stock options using the Black-Scholes valuation model. The Black-Scholes model requires the input of highly subjective assumptions. These assumptions include:
Expected Term. The expected term is estimated using the simplified method whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the option.
Volatility. Volatility is based on the historical trading volatility of our stock on the date of grant for a period consistent with the expected term.
Risk-Free Interest Rate. The risk-free interest rate is based on the zero-coupon U.S. Treasury instruments on the date of grant with a maturity date consistent with the expected term of our stock option grants.
Expected Dividend. To date, we have not declared or paid any cash dividends and do not have any plans to do so in the future. Therefore, we use an expected dividend yield of zero.
As required, we review our valuation assumptions at each grant date and, as a result, we may change our valuation assumptions used to value employee stock-based awards granted in future periods. Employee and director stock-based compensation costs are recognized over the vesting period of the award.
Concentration of Credit Risk
ASC 825, “Financial Instruments,” requires disclosure of any significant off‑balance sheet risk and credit risk concentration. We do not have significant off‑balance sheet risk or credit concentration. We maintain cash and short-term investments with major financial institutions. From time to time we have funds on deposit with commercial banks that exceed federally insured limits. The balances are insured by the Federal Deposit Insurance Corporation up to $250,000. At December 31, 2017 our uninsured cash balance was $8,399,154. Management does not consider this to be a significant credit risk as the banks are large, established financial institutions.
Recently Issued Accounting Standards
See Note 2, “Summary of Significant Accounting Policies in the Notes to the Financial Statements,” in the Notes to Financial Statements in this Annual Report for a discussion of recent accounting pronouncements.
Results of Operations
Comparison of the Years Ended December 31, 2017 and December 31, 2016
Total Revenues
We had no revenues for the years ended December 31, 2017 or 2016.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expenses for executive, finance and other administrative personnel, recruitment expenses, professional fees and other corporate expenses, including business development, investor relations, and general legal activities.
General and administrative expenses increased approximately $315,000, or 5.0%, to $6,639,000 for the year ended December 31, 2017 from $6,324,000 for the year ended December 31, 2016. The year over year increase is primarily attributable to an increase in personnel expenses and professional fees.
Research and Development Expenses
Research and development expenses increased approximately $626,000, or 6.2%, to $10,715,000 for the year ended December 31, 2017, from $10,089,000 for the year ended December 31, 2016. The increase in research and development costs is primarily attributable to increased clinical trial costs related to the progression of our Phase 2a proof-of-concept clinical trials for RX-3117, which we are currently evaluating in patients with relapsed or refractory metastatic pancreatic cancer and locally advanced or metastatic bladder cancer.
The table below summarizes the approximate amounts incurred on each of our research and development projects for the years ended December 31, 2017 and 2016:
|
For the Year Ended December 31,
|
||||||||
|
2017
|
2016
|
|||||||
|
Clinical Candidates:
|
||||||||
|
RX-3117
|
$
|
4,559,200
|
$
|
2,290,000
|
||||
|
RX-5902
|
2,019,700
|
2,230,800
|
||||||
|
RX-0201
|
535,700
|
1,573,800
|
||||||
|
Preclinical, Personnel and Overhead
|
3,600,696
|
3,994,549
|
||||||
|
Total Research and Development Expenses
|
$
|
10,715,296
|
$
|
10,089,149
|
||||
Interest Income
Interest income increased approximately $88,000, or 74.6% to $207,000 for the year ended December 31, 2017 from $119,000 for the year ended December 31, 2016. The increase is primarily attributable to higher interest rates and larger balances on cash and cash equivalents, and marketable securities for the year ended December 31, 2017 compared to the year ended December 31, 2016.
Mediation Settlement
During the year ended December 31, 2016, we received approximately $1,771,000 from a binding, one-time settlement agreement with one of our Japanese patent attorneys in exchange for our agreement not to bring any future claims related to a patent filing in Japan.
Unrealized (Loss) Gain on Fair Value of Warrants
Our warrants are recorded as liabilities at fair value, and the warrants are valued using a lattice model. Changes in the fair value of warrants are recorded as an unrealized gain or loss in our statement of operations. During the years ended December 31, 2017 and 2016, we recorded unrealized (losses) gains on the fair value of warrants of approximately ($7,594,000) and $5,530,000 respectively. Estimating fair values of warrants requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the warrants due to related changes to external market factors. The large unrealized loss for the year ended December 31, 2017 primarily resulted from a significant increase in the stock price of the underlying common stock on December 31, 2017, as compared to December 31, 2016 and from the price of the common stock compared to the warrant exercise price on dates during the year when warrants were exercised. An increase in volatility of the common stock and an increase in the number of outstanding warrants at times during the year ended December 31, 2017 also impacted the large unrealized loss for that period.
Financing Expense
We incurred approximately $553,000 and $313,000 of financing expenses during the year ended December 31, 2017, and 2016, respectively related to our registered direct offerings in October 2017, June 2017, September 2016 and March 2016.
Net Loss
Net loss for the year ended December 31, 2017 increased approximately $15,988,000 or 171.8%, to $25,295,000 ($0.92 per share) from $9,307,000 ($0.43 per share) for the year ended December 31, 2016, primarily as a result of the change from an unrealized gain on the fair value of warrants in 2016 to an unrealized loss on the fair value of warrants in 2017.
Comparison of the Years Ended December 31, 2016 and December 31, 2015
Total Revenues
We had no revenues for the years ended December 31, 2016 or 2015.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expenses for executive, finance and other administrative personnel, recruitment expenses, professional fees and other corporate expenses, including business development, investor relations, and general legal activities.
General and administrative expenses increased approximately $209,000, or 3.4%, to $6,324,000 for the year ended December 31, 2016 from $6,115,000 for the year ended December 31, 2015. The year over year increase is primarily attributable to an increase in personnel expenses.
Research and Development Expenses
Research and development expenses decreased approximately $2,059,000, or 16.9%, to $10,089,000 for the year ended December 31, 2016, from $12,148,000 for the year ended December 31, 2015. Decreased research and development costs for the year ended December 31, 2016 were primarily attributable to lower manufacturing costs for our drug candidates due to a significant supply of our drug candidates already being available to us from earlier manufacturing campaigns. During the year ended December 31, 2016, we incurred approximately $2,564,000 of drug manufacturing costs, compared to approximately $5,614,000 during the year ended December 31, 2015. Because the volume and timing of drug manufacturing does not correlate directly with the level and timing of clinical trial activity, we expect expenses related to drug manufacturing costs to vary from period to period based not only on the progress of clinical trials, but also when we engage in manufacturing activities. The decreases to drug manufacturing costs were partially offset by increases in clinical costs related to patient and site enrollment and personnel costs.
The table below summarizes the approximate amounts incurred on each of our research and development projects for the years ended December 31, 2016 and 2015:
|
For the Year Ended December 31,
|
||||||||
|
2016
|
2015
|
|||||||
|
Clinical Candidates:
|
||||||||
|
RX-3117
|
$
|
2,290,000
|
$
|
4,062,000
|
||||
|
RX-5902
|
2,230,800
|
2,839,000
|
||||||
|
RX-0201
|
1,573,800
|
1,547,000
|
||||||
|
Preclinical, Personnel and Overhead
|
3,994,549
|
3,700,226
|
||||||
|
Total Research and Development Expenses
|
$
|
10,089,149
|
$
|
12,148,226
|
||||
Interest Income
Interest income increased approximately $16,000 or 14.8% to $119,000 for the year ended December 31, 2016 from $103,000 for the year ended December 31, 2015. The increase is primarily attributable to higher interest rates on cash and cash equivalents, and marketable securities for the year ended December 31, 2016 compared to the year ended December 31, 2015.
Mediation Settlement
During the year ended December 31, 2016, we received approximately $1,771,000 from a binding, one-time settlement agreement with one of our Japanese patent attorneys in exchange for our agreement not to bring any future claims related to a patent filing in Japan.
Unrealized Gain on Fair Value of Warrants
Our warrants are recorded as liabilities at fair value, and the warrants are valued using a lattice model. Changes in the fair value of warrants are recorded as an unrealized gain or loss in our statement of operations. During the years ended December 31, 2016 and 2015, we recorded unrealized gains on the fair value of our warrants of approximately $5,530,000 and $3,987,000 respectively. Estimating fair values of warrants requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the warrant with related changes to external market factors. The unrealized gains for the years ended December 31, 2016 and 2015 primarily resulted from a decreased stock price underlying the common stock at December 31, 2016 and 2015, and from the greater number of warrants outstanding in 2016 compared to 2015.
Financing Expense
We incurred approximately $313,000 and $211,000 of financing expenses during the years ended December 31, 2016 and 2015, respectively, related to our registered direct offerings in September 2016, March 2016, and November 2015.
Net Loss
As a result of the above, net loss for the years ended December 31, 2016 and 2015 was approximately $9,307,000 and $14,385,000 or $0.43 and $0.79 per share, respectively.
Research and Development Projects
Research and development costs are expensed as incurred. These costs consist primarily of salaries and related personnel costs, costs to acquire pharmaceutical products and product rights for development and amounts paid to CROs, hospitals and laboratories for the provision of services and materials for drug development and clinical trials. Costs incurred in obtaining the license rights to technology in the research and development stage that have no alternative future uses are expensed as incurred. Our research and development programs are related to our oncology drug candidates. As we expand our clinical studies, we expect to enter into additional development agreements. Significant additional expenditures will be required if we complete our clinical trials, start new trials, apply for regulatory approvals, continue development of our technologies, expand our operations and bring our products to market. The eventual total cost of each clinical trial is dependent on a number of uncertainties such as trial design, the length of the trial, the number of clinical sites and the number of patients. The process of obtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive and uncertain. Because the successful development of our most advanced drug candidates, RX-3117 and RX-5902 is uncertain, we are unable to estimate the costs of completing our research and development programs, the timing of bringing such programs to market and, therefore, when material cash inflows could commence from the sale of these drug candidates, if any. If these projects are not completed as planned, our results of operations and financial condition would be negatively affected.
RX-3117
RX-3117 is a novel, investigational oral small molecule nucleoside compound. We believe RX-3117 has therapeutic potential in a broad range of cancers including pancreatic, bladder, lung, cervical, non-small cell lung cancer and colon cancer. Additional information about RX-3117, including about the current Phase 2a clinical trials, can be found in Item 1 of this Annual Report. We expect that expenses related to RX-3117 will increase in 2018 compared to 2017 as we continue patient enrollment for clinical trials, including our Phase 2a clinical study of RX-3117 in combination with Abraxane® in patients newly diagnosed with metastatic pancreatic cancer, which began in November 2017, as well as for continued manufacturing costs for new campaigns.
RX-5902 (Supinoxin)
RX-5902 is a potential first-in-class small molecule inhibitor of phosphorylated p68, a protein that we believe plays a key role in cancer growth, progression and metastasis. Phosphorylated p68 results in up-regulation of cancer-related genes and a subsequent proliferation of cancer cells and tumor growth. Additional information about RX-5902, including about the Phase 2a clinical trial in cancer patients with triple negative breast cancer can be found in Item 1 of this Annual Report. We expect that expenses related to RX-5902 will increase in 2018 compared to 2017 as we continue our Phase 2a study in patients with triple negative breast cancer, and due to increased manufacturing costs for new campaigns.
RX-0201 (Archexin)
RX-0201 is a potential best-in-class, potent inhibitor of the protein kinase Akt-1, which we believe plays a critical role in cancer cell proliferation, survival, angiogenesis, metastasis and drug resistance. Additional information about RX-0201, including about our research collaboration with Zhejiang Haichang Biotechnology Co., Ltd., can be found in Item 1 to this Annual Report. We expect that expenses related to RX-0201 will decrease in 2018 compared to 2017 as we wind down our Phase 2a clinical trial of RX-0201 in patients with metastatic renal cell carcinoma.
Pre-clinical Pipeline
We expect that expenses related to our pre-clinical pipeline, will remain flat in 2018 compared to 2017 as we continue testing and development.
Research and Development Process
We have engaged third-party CROs and other investigators and collaborators, such as universities, medical institutions and other life science companies, to conduct our pre-clinical studies, toxicology studies and clinical trials. Engaging third parties is typical practice in our industry. However, relying on such organizations means that the clinical trials and other studies described above are being conducted at external locations and the completion of these trials and studies is not within our direct control. Trials and studies may be delayed due to circumstances outside our control, and such delays may result in additional expenses for us.
Liquidity and Capital Resources
Cash Flows
The table below summarizes our net cash flow activity:
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Net Cash Used in Operating Activities
|
$
|
(15,420,055
|
)
|
$
|
(13,227,101
|
)
|
$
|
(17,351,950
|
)
|
|||
|
Net Cash (Used In) Provided by Investing Activities
|
(9,372,778
|
)
|
4,483,911
|
9,554,394
|
||||||||
|
Net Cash Provided by Financing Activities
|
22,113,514
|
10,122,223
|
8,170,751
|
|||||||||
|
Net (Decrease) Increase in Cash and Cash Equivalents
|
$
|
(2,679,319
|
)
|
$
|
1,379,033
|
$
|
373,195
|
|||||
Cash used in operating activities was approximately $15,420,000 for the year ended December 31, 2017. The operating cash flows during the year ended December 31, 2017 reflect a net loss of $25,295,000 offset by an unrealized loss on the fair value of warrants of $7,594,000 and a net increase of cash components of working capital and non-cash charges totaling $2,281,000. Cash used in operating activities was approximately $13,227,000 for the year ended December 31, 2016. The operating cash flows during the year ended December 31, 2016 reflect our net loss of $9,307,000, an unrealized gain on the fair value of warrants of $5,530,000 and a net increase of cash components of working capital and non-cash charges totaling $1,610,000. Cash used in operating activities was approximately $17,352,000 for the year ended December 31, 2015. The operating cash flows during the year ended December 31, 2015 reflect our net loss of $14,385,000, an unrealized gain on the fair value of warrants of $3,987,000 and a net increase of cash components of working capital and non-cash charges totaling $1,020,000.
Cash used in investing activities was approximately $9,373,000 for the year ended December 31, 2017, which consisted of $21,018,000 and $75,000 for purchases of marketable securities and equipment, respectively, offset by $11,720,000 from the redemption of marketable securities. Cash provided by investing activities was approximately $4,484,000 for the year ended December 31, 2016, which consisted of $13,240,000 from the redemption of marketable securities, offset by $8,747,000 and $9,000 for the purchases of marketable securities and equipment, respectively. Cash provided by investing activities was approximately $9,554,000 for the year ended December 31, 2015, which consisted of $17,525,000 from the redemption of marketable securities, offset by $7,909,000 and $62,000 for the purchases of marketable securities and equipment, respectively.
Cash provided by financing activities was approximately $22,114,000 for the year ended December 31, 2017, which consisted of net proceeds of $16,682,000 from our registered direct public offerings in June 2017 and October 2017, and $5,354,000 and $78,000 from the exercise of stock warrants and options, respectively. Cash provided by financing activities was approximately $10,122,000 for the year ended December 31, 2016 which consisted of net proceeds from our registered direct public offerings in March 2016 and September 2016. Cash provided by financing activities was approximately $8,171,000 for the year ended December 31, 2015, which consisted of net proceeds of $7,440,000 from our registered direct public offering in November 2015 and sales from our at market issuance agreement, and proceeds of $709,000 and $22,000 received from the exercise of stock options and stock warrants, respectively.
Financings
On November 12, 2015, we closed a registered direct public offering of 1,666,667 shares of common stock and warrants to purchase up to 1,250,000 shares of common stock. The common stock and warrants were sold in units consisting of one share of common stock and a warrant to purchase 0.75 shares of common stock at a price of $4.20 per unit, and the warrants have an exercise price of $5.30 per share. The total gross proceeds of the offering were $7,000,000. The warrants issued are exercisable beginning six months after the closing date until the five-year anniversary of the initial exercise date.
On March 2, 2016, we closed a registered direct public offering of 1,562,500 shares of common stock and warrants to purchase up to 1,171,875 shares of common stock. The common stock and warrants were sold in units consisting of one share of common stock and a warrant to purchase 0.75 shares of common stock at a price of $3.20 per unit, and the warrants have an exercise price of $4.20 per share. The total gross proceeds of the offering were $5,000,000. The warrants are exercisable beginning six months after the closing date until the five-year anniversary of the initial exercise date.
On September 19, 2016, we closed a registered direct public offering of 2,400,000 shares of common stock and warrants to purchase up to 1,800,000 shares of common stock. The common stock and warrants were sold in units consisting of one share of common stock and a warrant to purchase 0.75 shares of common stock at a price of $2.50 per unit, and the warrants have an exercise price of $3.00 per share. The total gross proceeds of the offering were $6,000,000. The warrants are exercisable beginning six months after the closing date until the five-year anniversary of the initial exercise date.
On June 12, 2017, we closed a registered direct public offering of 3,030,304 shares of common stock and warrants to purchase up to 1,515,152 shares of common stock. The common stock and warrants were sold in units consisting of one share of common stock and a warrant to purchase 0.5 shares of common stock at a price of $3.30 per unit, and the warrants have an exercise price of $4.00 per share. The total gross proceeds of the offering were $10,000,003. The warrants are exercisable beginning six months after the closing date until the five-year anniversary of the initial exercise date.
On October 17, 2017, we closed a registered direct public offering of 3,265,309 shares of common stock and warrants to purchase up to 1,632,654 shares of common stock. The common stock and warrants were sold in units consisting of one share of common stock and a warrant to purchase 0.5 shares of common stock at a price of $2.45 per unit, and the warrants have an exercise price of $2.85 per share. The total gross proceeds of the offering were $8,000,007. The warrants are exercisable beginning six months after the closing date until the five-year anniversary of the initial exercise date.
We will need to raise additional capital through public or private equity or debt offerings or through arrangements with strategic partners or other sources in order to continue to develop our drug candidates. There can be no assurance that additional capital will be available when needed or on terms satisfactory to us, if at all. If we are not able to raise sufficient additional capital, we will have to reduce our research and development activities. We will first reduce research and development activities associated with our pre-clinical compounds. To the extent necessary, we will then reduce our research and development activities related to some or all of our clinical drugs.
At Market Issuance Sales Agreement
On August 2, 2017, we terminated the at market issuance sales agreement, dated as of March 16, 2015, with MLV & Co. LLC, now part of FBR & Co. (“MLV”), pursuant to which we were entitled to issue and sell shares of our common stock having an aggregate offering price of up to $40 million from time to time, at our option, through MLV as our sales agent. For the year ended December 31, 2015, we sold 140,707 shares of common stock pursuant to the sales agreement for $1,042,573 in gross proceeds. There were no sales under the sales agreement in 2016 or 2017.
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2017:
|
Total
|
Less than 1
year
|
1 -3 Years
|
3-5 Years
|
More than 5
years
|
||||||||||||||||
|
Operating Leases
|
$
|
489,822
|
$
|
279,274
|
$
|
210,548
|
$
|
-
|
$
|
-
|
||||||||||
We also have obligations under various license agreements that become due and payable on the achievement of certain development, regulatory, or commercial milestones. We have not included these commitments on our balance sheet or in the above table of contractual obligations because the achievement and timing of these events is neither fixed nor determinable.
We have contracted with various vendors for research and development services, the terms of which require payments over the term of the agreements, usually ranging from two to 36 months. The costs to be incurred are estimated and are subject to revision. As of December 31, 2017, the total estimated cost to complete these agreements was approximately $11,110,000. All of these agreements may be terminated by either party upon appropriate notice as stipulated in the respective agreements, and therefore, are not included in the above table of contractual obligations.
Current and Future Financing Needs
We have incurred negative cash flow from operations since we started our business. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our planned product development efforts, our clinical trials and our research and development efforts. We will need to raise additional capital through public or private equity or debt offerings or through arrangements with strategic partners or other sources in order to continue to develop our drug candidates. There can be no assurance that additional capital will be available when needed or on terms satisfactory to us, if at all. If we are not able to raise sufficient additional capital, we will have to reduce our research and development activities. We believe our cash, cash equivalents, and marketable securities will be sufficient to cover our cash flow requirements for our current activities for at least the next 12 months from the date our financial statements are issued.
The actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
| · |
the progress of our product development activities;
|
| · |
the number and scope of our product development programs;
|
| · |
the progress of our pre-clinical and clinical trial activities;
|
| · |
the progress of the development efforts of parties with whom we have entered into collaboration agreements;
|
| · |
our ability to maintain current collaboration programs and to establish new collaboration arrangements;
|
| · |
the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; and
|
| · |
the costs and timing of regulatory approvals.
|
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or holdings in variable interest entities.
For the year ended December 31, 2017, we are exposed to the following market risks:
Interest Rate Risk
We invest our cash in a variety of financial instruments. At December 31, 2017, our cash and cash equivalents were invested primarily in short term bank deposits and municipal obligations, all of which were denominated in U.S. dollars. Due to the conservative nature of these investments, which primarily bear interest at fixed rates, we do not believe we have material exposure to interest rate risk.
Foreign Currency Risk
We are exposed to risks associated with foreign currency transactions on contracts with vendors associated outside of the United States. Accordingly changes in the value of the U.S. dollar, relative to other currencies, may have an impact on our financial statements and earnings. The number and dollar amount of contracts denominated in foreign currency is immaterial; therefore, we believe we do not have material exposure to foreign currency risk.
Our financial statements and the Report of the Independent Registered Public Accounting Firm thereon filed pursuant to this Item 8 and are included in this Annual Report beginning on page F-1.
None.
Evaluation of Disclosure Controls and Procedures. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this report. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective such that the information required to be disclosed by us in reports filed under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding disclosure. A controls system cannot provide absolute assurance, however, that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
Changes in Internal Control Over Financial Reporting. During the most recent quarter ended December 31, 2017, there has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act) that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles and includes those policies and procedures that:
| · |
pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and the dispositions of our assets;
|
| · |
provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and the board of directors; and
|
| · |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements.
|
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluations of effectiveness to future periods are subject to risk that controls may become inadequate because of changes in conditions or because of declines in the degree of compliance with the policies or procedures.
Our management, with the participation of the Chief Executive Officer and Chief Financial Officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in the Internal Control-Integrated Framework (2013).
Based on this evaluation, our management, with the participation of the Chief Executive Officer and Chief Financial Officer, concluded that, as of December 31, 2017, our internal control over financial reporting was effective.
Management’s assessment of the effectiveness of our internal control over financial reporting has been audited by Baker Tilly Virchow Krause, LLP, an independent registered public accounting firm. Baker Tilly Virchow Krause, LLP has issued an attestation report on the effectiveness of our internal control over financial reporting, which appears in Item 8 of this Annual Report.
None.
PART III
The information required by this Item is set forth in our 2018 Proxy Statement to be filed with the SEC within 120 days of December 31, 2017 and is incorporated into this Annual Report by reference.
The information required by this Item is set forth in our 2018 Proxy Statement to be filed with the SEC within 120 days of December 31, 2017 and is incorporated into this Annual Report by reference.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
|
The information required by this Item is set forth in our 2018 Proxy Statement to be filed with the SEC within 120 days of December 31, 2017 and is incorporated into this Annual Report by reference.
The information required by this Item is set forth in our 2018 Proxy Statement to be filed with the SEC within 120 days of December 31, 2017 and is incorporated into this Annual Report by reference.
The information required by this Item is set forth in our 2018 Proxy Statement to be filed with the SEC within 120 days of December 31, 2017 and is incorporated into this Annual Report by reference.
| (a) |
The following documents are filed as a part of this Annual Report:
|
|
(1)
|
The following documents are filed as a part of this Annual Report:
|
||
|
Report of Baker Tilly Virchow Krause, LLP
|
F-1
|
||
|
Balance Sheet as of December 31, 2017 and December 31, 2016
|
F-3
|
||
|
Statement of Operations for the year ended December 31, 2017, 2016 and 2015
|
F-4
|
||
|
Statement of Comprehensive Loss for the year ended December 31, 2017, 2016 and 2015
|
F-5
|
||
|
Statement of Stockholders’ Equity for the year ended December 31, 2017, 2016 and 2015
|
F-6
|
||
|
Statement of Cash Flows for the year ended December 31, 2017, 2016 and 2015
|
F-7
|
||
|
Notes to the Financial Statements
|
F-8
|
||
|
(2)
|
All financial statement schedules have been omitted because they are not applicable or not required or because the information is included elsewhere in the financial statements or the Notes thereto.
|
||
|
(3)
|
See the accompanying Index to Exhibits filed as a part of this Annual Report, which list is incorporated by reference in this Item.
|
| (b) |
See the accompanying Index to Exhibits filed as a part of this Annual Report.
|
| (c) |
Other schedules are not applicable.
|
None.
INDEX TO EXHIBITS
|
Amended and Restated Certificate of Incorporation, filed as Appendix G to the Company’s Definitive Proxy Statement on Schedule 14A (File No. 000-50590) dated April 29, 2005, is incorporated herein by reference.
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation, filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed May 5, 2017, is incorporated herein by reference.
|
|
|
Amended and Restated Bylaws, as amended, through March 21, 2014, filed as Exhibit 3.2 to the Company’s Annual Report on Form 10-K on March 21, 2014, is incorporated herein by reference.
|
|
|
Specimen Certificate for the Company’s Common Stock, par value $.0001 per share, filed as Exhibit 4.3 to the Company’s Registration Statement on Form S-8 (File No. 333-129294) dated October 28, 2005, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on July 24, 2013, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on October 16, 2013, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on January 15, 2014, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on November 6, 2015, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on February 26, 2016, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on September 14, 2016, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on June 7, 2017, is incorporated herein by reference.
|
|
|
Form of Warrant for the Company’s Common Stock Purchase Warrants, filed as Exhibit 4.1 to the Company’s Current Report on Form 8-K filed on October 13, 2017, is incorporated herein by reference.
|
|
|
Rexahn Pharmaceuticals, Inc. Stock Option Plan, as amended, filed as Exhibit 4.4 to the Company’s Registration Statement on Form S-8 (File No. 333-129294) dated October 28, 2005, is incorporated herein by reference.
|
|
|
Form of Stock Option Grant Agreement for Employees, filed as Exhibit 4.5.1 to the Company’s Registration Statement on Form S-8 (File No. 333-129294) dated October 28, 2005, is incorporated herein by reference.
|
|
Form of Stock Option Grant Agreement for Non-Employee Directors and Consultants, filed as Exhibit 4.5.2 to the Company’s Registration Statement on Form S-8 (File No. 333-129294) dated October 28, 2005, is incorporated herein by reference.
|
|
|
Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan, as amended and restated, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 10, 2016, is incorporated herein by reference.
|
|
|
First Amendment to the Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan, as amended and restated as of June 9, 2016, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 13, 2017, is incorporated herein by reference.
|
|
|
Form of Stock Option Grant Agreement under Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan filed as Exhibit 10.5 to the Company’s Annual Report on Form 10-K for the year ended December 31, 2015, filed on March 14, 2016, is incorporated herein by reference.
|
|
|
Employment Agreement, dated as of September 9, 2010, by and between Rexahn Pharmaceuticals, Inc. and T. H. Jeong, filed as Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on September 10, 2010, is incorporated herein by reference.
|
|
|
Separation, Transition and General Release Agreement, dated as of December 11, 2017, by and between Rexahn Pharmaceuticals, Inc. and Tae Heum (Ted) Jeong.
|
|
|
Employment Agreement, dated as of February 4, 2013, by and between Rexahn Pharmaceuticals, Inc. and Peter Suzdak, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on January 22, 2013, is incorporated herein by reference.
|
|
|
Employment Agreement, dated as of February 2, 2015, by and between Rexahn Pharmaceuticals, Inc. and Ely Benaim, M.D., filed as Exhibit 10.1 to the Company’s Current Report on Form 10-Q filed on May 8, 2015, is incorporated herein by reference.
|
|
|
Bonus Letter Agreement, dated as of August 2, 2016, by and between Rexahn Pharmaceuticals, Inc. and Ely Benaim, M.D., filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2016, is incorporated herein by reference.
|
|
|
Employment Agreement, dated as of July 6, 2016, by and between Rexahn Pharmaceuticals, Inc. and Lisa Nolan, Ph.D., filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2016, is incorporated herein by reference.
|
|
|
Employment Agreement, dated as of January 2, 2018, by and between Rexahn Pharmaceuticals, Inc. and Douglas Swirsky, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed January 4, 2018 is incorporated herein by reference.
|
|
|
Lease Agreement, dated June 5, 2009, by and between Rexahn Pharmaceuticals, Inc. and The Realty Associates Fund V, L.P., filed as Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2009, is incorporated herein by reference.
|
|
|
First Amendment to Lease Agreement, dated as of June 7, 2013, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2013, is incorporated herein by reference.
|
|
Second Amendment to Lease Agreement, dated as of July 26, 2014, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2014, is incorporated herein by reference.
|
|
|
Third Amendment to Lease Agreement, dated as of May 6, 2015, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2015, is incorporated herein by reference.
|
|
|
Fourth Amendment to Lease Agreement, dated as of April 4, 2016, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC, filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2016, is incorporated herein by reference.
|
|
|
Fifth Amendment to Lease Agreement, dated as of April 13, 2017, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC, filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2017, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of July 23, 2013, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 24, 2013, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of October 10, 2013, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 16, 2013, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of January 15, 2014, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on January 15, 2014, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of November 6, 2015, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 6, 2015, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of February 26, 2016, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 26, 2016, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of September 14, 2016, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on September 14, 2016, is incorporated herein by reference.
|
|
|
Form of Securities Purchase Agreement, dated as of June 6, 2017, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 7, 2017, is incorporated herein by reference.
|
|
Form of Securities Purchase Agreement, dated as of October 13, 2017, by and between Rexahn Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto, filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 13, 2017, is incorporated herein by reference.
|
|
|
Statement Regarding the Computation of Ratio of Earnings to Combined Fixed Charges and Preferred Stock Dividends
|
|
|
Consent of Baker Tilly Virchow Krause, LLP, independent registered public accounting firm
|
|
|
Power of Attorney
|
|
|
Certification of Chief Executive Officer Pursuant to Rule 13a-14(a) or Rule 15d-14(a)
|
|
|
Certification of Chief Financial Officer Pursuant to Rule 13a-14(a) or Rule 15d-14(a)
|
|
|
Certification of Chief Executive Officer of Periodic Report Pursuant to 18 U.S.C. Section 1350
|
|
|
Certification of Chief Financial Officer of Periodic Report Pursuant to 18 U.S.C. Section 1350
|
|
|
101.INS
|
XBRL Instance Document
|
|
101.SCH
|
XBRL Taxonomy Extension Schema
|
|
101.CAL
|
XBRL Taxonomy Calculation Linkbase
|
|
101.DEF
|
XBRL Taxonomy Definition Linkbase
|
|
101.LAB
|
XBRL Taxonomy Label Linkbase
|
|
101.PRE
|
XBRL Taxonomy Presentation Linkbase
|
*Indicates management contract or compensatory plan or arrangement
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
REXAHN PHARMACEUTICALS, INC.
|
|||
|
By:
|
/s/ Douglas J. Swirsky
|
||
|
Douglas J. Swirsky
|
|||
|
Chief Financial Officer and President
|
|||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
|
Name
|
Title
|
Date
|
|
|
/s/ Peter D. Suzdak*
|
Chief Executive Officer and
|
March 9, 2018
|
|
|
Peter Suzdak
|
Director (Principal Executive Officer)
|
||
|
/s/ Douglas J. Swirsky
|
Chief Financial Officer and
|
March 9, 2018
|
|
|
Douglas J. Swirsky
|
President (Principal Financial and Accounting Officer)
|
||
|
/s/ Peter Brandt*
|
Chairman
|
March 9, 2018
|
|
|
Peter Brandt
|
|||
|
/s/ Charles Beever*
|
Director
|
March 9, 2018
|
|
|
Charles Beever
|
|||
|
/s/ Kwang Soo Cheong*
|
Director
|
March 9, 2018
|
|
|
Kwang Soo Cheong
|
|||
|
/s/ Mark Carthy*
|
Director
|
March 9, 2018
|
|
|
Mark Carthy
|
|||
|
/s/ Richard J. Rodgers*
|
Director
|
March 9, 2018
|
|
|
Richard J. Rodgers
|
* By: /s/ Douglas J. Swirsky, Attorney-in Fact
Douglas J. Swirsky, Attorney-in-Fact**
** By authority of the power of attorney filed as Exhibit 24.1 hereto
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the stockholders and the board of directors of Rexahn Pharmaceuticals, Inc.:
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying balance sheet of Rexahn Pharmaceuticals, Inc. (the "Company") as of December 31, 2017 and 2016, the related statements of operations, comprehensive loss, stockholders’ equity and cash flows, for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the "financial statements"). We also have audited the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control –Integrated Framework: (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework: (2013) issued by COSO.
Basis for Opinions
The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company's financial statements and an opinion on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the financial statements included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control Over Financial Reporting
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Baker Tilly Virchow Krause, LLP
We are uncertain as to the year we (or our predecessor firms) began serving consecutively as the auditor of the Company’s financial statements; however, we are aware that we (or our predecessor firms) have been have been the Company’s auditor consecutively since at least 2003.
Wyomissing, Pennsylvania
March 9, 2018
REXAHN PHARMACEUTICALS, INC.
Balance Sheet
|
December 31, 2017
|
December 31, 2016
|
|||||||
|
ASSETS
|
||||||||
|
Current Assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
8,899,154
|
$
|
11,578,473
|
||||
|
Marketable securities
|
17,931,941
|
8,737,107
|
||||||
|
Prepaid expenses and other current assets
|
1,304,541
|
608,517
|
||||||
|
Total Current Assets
|
28,135,636
|
20,924,097
|
||||||
|
Security Deposits
|
30,785
|
30,785
|
||||||
|
Equipment, Net
|
121,460
|
88,650
|
||||||
|
Total Assets
|
$
|
28,287,881
|
$
|
21,043,532
|
||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current Liabilities:
|
||||||||
|
Accounts payable and accrued expenses
|
$
|
3,233,926
|
$
|
1,882,500
|
||||
|
Deferred Research and Development Arrangement
|
375,000
|
450,000
|
||||||
|
Other Liabilities
|
56,724
|
79,204
|
||||||
|
Warrant Liabilities
|
7,853,635
|
1,573,366
|
||||||
|
Total Liabilities
|
11,519,285
|
3,985,070
|
||||||
|
Commitments and Contingencies (note 15)
|
||||||||
|
Stockholders’ Equity:
|
||||||||
|
Preferred stock, par value $0.0001, 10,000,000 authorized shares, none issued and outstanding
|
-
|
-
|
||||||
|
Common stock, par value $0.0001, 50,000,000 authorized shares, 31,725,114 and 23,736,878 issued and outstanding
|
3,173
|
2,374
|
||||||
|
Additional paid-in capital
|
157,141,021
|
132,086,419
|
||||||
|
Accumulated other comprehensive loss
|
(56,886
|
)
|
(6,122
|
)
|
||||
|
Accumulated deficit
|
(140,318,712
|
)
|
(115,024,209
|
)
|
||||
|
Total Stockholders’ Equity
|
16,768,596
|
17,058,462
|
||||||
|
Total Liabilities and Stockholders’ Equity
|
$
|
28,287,881
|
$
|
21,043,532
|
||||
(See accompanying notes to the financial statements)
REXAHN PHARMACEUTICALS, INC.
Statement of Operations
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Revenues:
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||
|
Expenses:
|
||||||||||||
|
General and administrative
|
6,639,421
|
6,324,236
|
6,115,210
|
|||||||||
|
Research and development
|
10,715,296
|
10,089,149
|
12,148,226
|
|||||||||
|
Total Expenses
|
17,354,717
|
16,413,385
|
18,263,436
|
|||||||||
|
Loss from Operations
|
(17,354,717
|
)
|
(16,413,385
|
)
|
(18,263,436
|
)
|
||||||
|
Other Income (Expense)
|
||||||||||||
|
Interest income
|
207,003
|
118,565
|
103,269
|
|||||||||
|
Mediation settlement
|
-
|
1,770,658
|
-
|
|||||||||
|
Unrealized (loss) gain on fair value of warrants
|
(7,594,162
|
)
|
5,529,907
|
3,986,727
|
||||||||
|
Financing expense
|
(552,627
|
)
|
(313,090
|
)
|
(211,116
|
)
|
||||||
|
Total Other Income (Expense)
|
(7,939,786
|
)
|
7,106,040
|
3,878,880
|
||||||||
|
Net Loss Before Provision for Income Taxes
|
(25,294,503
|
)
|
(9,307,345
|
)
|
(14,384,556
|
)
|
||||||
|
Provision for income taxes
|
-
|
-
|
-
|
|||||||||
|
Net Loss
|
$
|
(25,294,503
|
)
|
$
|
(9,307,345
|
)
|
$
|
(14,384,556
|
)
|
|||
|
Net loss per share, basic and diluted
|
$
|
(0.92
|
)
|
$
|
(0.43
|
)
|
$
|
(0.79
|
)
|
|||
|
Weighted average number of shares outstanding, basic and diluted
|
27,390,527
|
21,744,740
|
18,238,822
|
|||||||||
(See accompanying notes to the financial statements)
REXAHN PHARMACEUTICALS, INC.
Statement of Comprehensive Loss
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Net Loss
|
$
|
(25,294,503
|
)
|
$
|
(9,307,345
|
)
|
$
|
(14,384,556
|
)
|
|||
|
Unrealized (loss) gain on available-for-sale securities
|
(50,764
|
)
|
11,919
|
15,606
|
||||||||
|
Comprehensive Loss
|
$
|
(25,345,267
|
)
|
$
|
(9,295,426
|
)
|
$
|
(14,368,950
|
)
|
|||
REXAHN PHARMACEUTICALS, INC.
Statement of Stockholders’ Equity
For the Year Ended December 31, 2017, 2016 and 2015
|
Common Stock
|
Treasury Stock
|
|||||||||||||||||||||||||||||||
|
Number of
Shares
|
Amount
|
Additional
Paid-in
Capital
|
Accumulated
Deficit
|
Number
of Shares
|
Amount
|
Accumulated
Other
Comprehensive
Loss
|
Total
Stockholders'
Equity
|
|||||||||||||||||||||||||
|
Balances at January 1, 2015
|
17,836,652
|
$
|
1,784
|
$
|
118,073,072
|
$
|
(91,332,308
|
)
|
11,321
|
$
|
(128,410
|
)
|
$
|
(33,647
|
)
|
$
|
26,580,491
|
|||||||||||||||
|
Issuance of common stock and units
|
1,807,374
|
181
|
5,249,892
|
-
|
-
|
-
|
-
|
5,250,073
|
||||||||||||||||||||||||
|
Stock issuance costs
|
-
|
-
|
(566,065
|
)
|
-
|
-
|
-
|
-
|
(566,065
|
)
|
||||||||||||||||||||||
|
Common stock issued in exchange for services
|
15,000
|
2
|
101,998
|
-
|
-
|
-
|
-
|
102,000
|
||||||||||||||||||||||||
|
Stock options exercised
|
88,943
|
9
|
708,608
|
-
|
-
|
-
|
-
|
708,617
|
||||||||||||||||||||||||
|
Stock warrants exercised
|
4,730
|
-
|
31,703
|
-
|
-
|
-
|
-
|
31,703
|
||||||||||||||||||||||||
|
Stock-based compensation
|
-
|
-
|
1,037,679
|
-
|
-
|
-
|
-
|
1,037,679
|
||||||||||||||||||||||||
|
Retirement of treasury stock
|
(11,321
|
)
|
(1
|
)
|
(128,409
|
)
|
-
|
(11,321
|
)
|
128,410
|
-
|
-
|
||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(14,384,556
|
)
|
-
|
-
|
-
|
(14,384,556
|
)
|
||||||||||||||||||||||
|
Other comprehensive income
|
-
|
-
|
-
|
-
|
-
|
-
|
15,606
|
15,606
|
||||||||||||||||||||||||
|
Balances at December 31, 2015
|
19,741,378
|
$
|
1,975
|
$
|
124,508,478
|
$
|
(105,716,864
|
)
|
-
|
$
|
-
|
$
|
(18,041
|
)
|
$
|
18,775,548
|
||||||||||||||||
|
Issuance of common stock and units
|
3,962,500
|
396
|
6,908,562
|
-
|
-
|
-
|
-
|
6,908,958
|
||||||||||||||||||||||||
|
Stock issuance costs
|
-
|
-
|
(837,755
|
)
|
-
|
-
|
-
|
-
|
(837,755
|
)
|
||||||||||||||||||||||
|
Common stock issued in exchange for services
|
33,000
|
3
|
97,646
|
-
|
-
|
-
|
-
|
97,649
|
||||||||||||||||||||||||
|
Stock-based compensation
|
-
|
-
|
1,409,488
|
-
|
-
|
-
|
-
|
1,409,488
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(9,307,345
|
)
|
-
|
-
|
-
|
(9,307,345
|
)
|
||||||||||||||||||||||
|
Other comprehensive income
|
-
|
-
|
-
|
-
|
-
|
-
|
11,919
|
11,919
|
||||||||||||||||||||||||
|
Balances at December 31, 2016
|
23,736,878
|
$
|
2,374
|
$
|
132,086,419
|
$
|
(115,024,209
|
)
|
-
|
$
|
-
|
$
|
(6,122
|
)
|
$
|
17,058,462
|
||||||||||||||||
|
Issuance of common stock and units
|
6,295,613
|
630
|
11,965,753
|
-
|
-
|
-
|
-
|
11,966,383
|
||||||||||||||||||||||||
|
Stock issuance costs
|
-
|
-
|
(1,470,536
|
)
|
-
|
-
|
-
|
-
|
(1,470,536
|
)
|
||||||||||||||||||||||
|
Common stock issued in exchange for services
|
15,000
|
2
|
31,198
|
-
|
-
|
-
|
-
|
31,200
|
||||||||||||||||||||||||
|
Stock-based compensation
|
-
|
-
|
1,044,167
|
-
|
-
|
-
|
-
|
1,044,167
|
||||||||||||||||||||||||
|
Stock options exercised
|
25,000
|
2
|
77,498
|
-
|
-
|
-
|
-
|
77,500
|
||||||||||||||||||||||||
|
Stock warrants exercised
|
1,652,623
|
165
|
13,406,522
|
-
|
-
|
-
|
-
|
13,406,687
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(25,294,503
|
)
|
-
|
-
|
-
|
(25,294,503
|
)
|
||||||||||||||||||||||
|
Other comprehensive loss
|
-
|
-
|
-
|
-
|
-
|
-
|
(50,764
|
)
|
(50,764
|
)
|
||||||||||||||||||||||
|
Balances at December 31, 2017
|
31,725,114
|
$
|
3,173
|
$
|
157,141,021
|
$
|
(140,318,712
|
)
|
-
|
$
|
-
|
$
|
(56,886
|
)
|
$
|
16,768,596
|
||||||||||||||||
(See accompanying notes to the financial statements)
REXAHN PHARMACEUTICALS, INC.
Statement of Cash Flows
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Cash Flows from Operating Activities:
|
||||||||||||
|
Net loss
|
$
|
(25,294,503
|
)
|
$
|
(9,307,345
|
)
|
$
|
(14,384,556
|
)
|
|||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Compensatory stock
|
31,200
|
97,649
|
102,000
|
|||||||||
|
Depreciation and amortization
|
42,358
|
32,916
|
27,498
|
|||||||||
|
Amortization of premiums and discounts on marketable securities, net
|
52,012
|
22,321
|
30,875
|
|||||||||
|
Stock-based compensation
|
1,044,167
|
1,409,488
|
1,037,679
|
|||||||||
|
Amortization of deferred research and development arrangement
|
(75,000
|
)
|
(75,000
|
)
|
(75,000
|
)
|
||||||
|
Unrealized loss (gain) on fair value of warrants
|
7,594,162
|
(5,529,907
|
)
|
(3,986,727
|
)
|
|||||||
|
Financing expense
|
552,627
|
313,090
|
211,116
|
|||||||||
|
Amortization of deferred lease incentive
|
(12,444
|
)
|
(12,443
|
)
|
(12,443
|
)
|
||||||
|
Deferred lease expenses
|
(10,036
|
)
|
(12,373
|
)
|
(8,492
|
)
|
||||||
|
Changes in assets and liabilities:
|
||||||||||||
|
Prepaid expenses and other assets
|
(696,024
|
)
|
613,301
|
(495,935
|
)
|
|||||||
|
Accounts payable and accrued expenses
|
1,351,426
|
(778,798
|
)
|
202,035
|
||||||||
|
Net Cash Used in Operating Activities
|
(15,420,055
|
)
|
(13,227,101
|
)
|
(17,351,950
|
)
|
||||||
|
Cash Flows from Investing Activities:
|
||||||||||||
|
Purchase of equipment
|
(75,168
|
)
|
(8,666
|
)
|
(62,302
|
)
|
||||||
|
Purchase of marketable securities
|
(21,017,610
|
)
|
(8,747,423
|
)
|
(7,908,304
|
)
|
||||||
|
Redemption of marketable securities
|
11,720,000
|
13,240,000
|
17,525,000
|
|||||||||
|
Net Cash (Used in) Provided by Investing Activities
|
(9,372,778
|
)
|
4,483,911
|
9,554,394
|
||||||||
|
Cash Flows from Financing Activities:
|
||||||||||||
|
Issuance of common stock and units, net of issuance costs
|
16,681,921
|
10,122,223
|
7,439,809
|
|||||||||
|
Proceeds from exercise of stock warrants
|
5,354,093
|
-
|
22,325
|
|||||||||
|
Proceeds from exercise of stock options
|
77,500
|
-
|
708,617
|
|||||||||
|
Net Cash Provided by Financing Activities
|
22,113,514
|
10,122,223
|
8,170,751
|
|||||||||
|
Net (Decrease) Increase in Cash and Cash Equivalents
|
(2,679,319
|
)
|
1,379,033
|
373,195
|
||||||||
|
Cash and Cash Equivalents – beginning of period
|
11,578,473
|
10,199,440
|
9,826,245
|
|||||||||
|
Cash and Cash Equivalents - end of period
|
$
|
8,899,154
|
$
|
11,578,473
|
$
|
10,199,440
|
||||||
|
Supplemental Cash Flow Information
|
||||||||||||
|
Non-cash financing and investing activities:
|
||||||||||||
|
Warrants issued
|
$
|
6,738,701
|
$
|
4,364,110
|
$
|
2,966,917
|
||||||
|
Warrant liability extinguishment from exercise of warrants
|
$
|
8,052,594
|
$
|
-
|
$
|
9,378
|
||||||
|
Retirement of treasury stock
|
$
|
-
|
$
|
-
|
$
|
128,410
|
||||||
(See accompanying notes to the financial statements)
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 1. |
Operations and Organization
|
Operations
Rexahn Pharmaceuticals, Inc. (the “Company”), a Delaware corporation, is a biopharmaceutical company whose principal operations are the discovery and development of innovative treatments for cancer. The Company had an accumulated deficit of $140,318,712 at December 31, 2017 and anticipates incurring losses through fiscal year 2018 and beyond. The Company has not yet generated commercial revenues and has funded its operating losses to date through the sale of shares of its common stock and warrants to purchase shares of its common stock, convertible debt, financings, interest income from cash, cash equivalents and marketable securities, and proceeds from reimbursed research and development costs. The Company believes that its cash, cash equivalents, and marketable securities, will be sufficient to cover its cash flow requirements for its current activities at least for the next 12 months from the date these financial statements were issued. Management believes it has the capability of managing the Company’s operations within existing cash available by focusing on select research and development activities, selecting projects in conjunction with potential financings and milestones, and efficiently managing its general and administrative affairs.
| 2. |
Summary of Significant Accounting Policies
|
a) Cash and Cash Equivalents
Cash and cash equivalents include cash on hand and short‑term investments purchased with remaining maturities of three months or less at acquisition.
b) Marketable Securities
Marketable securities are considered “available-for-sale” in accordance with Financial Statement Accounting Board (“FASB”) Accounting Standards Codification (“ASC”) 320, “Debt and Equity Securities,” and thus are reported at fair value in the Company’s accompanying balance sheet, with unrealized gains and losses excluded from earnings and reported as a separate component of stockholders’ equity. Amounts reclassified out of accumulated other comprehensive loss into realized gains and losses are accounted for on the basis of specific identification and are included in other income or expense in the statement of operations. The Company classifies such investments as current on the balance sheet as the investments are readily marketable and available for use in the Company’s current operations.
c) Equipment
Equipment is stated at cost less accumulated depreciation. Depreciation, based on the lesser of the term of the lease or the estimated useful life of the assets, is provided as follows:
|
Life
|
Depreciation Method
|
||
|
Furniture and fixtures
|
7 years
|
straight line
|
|
|
Office equipment
|
5 years
|
straight line
|
|
|
Lab equipment
|
5-7 years
|
straight line
|
|
|
Computer equipment
|
3-5 years
|
straight line
|
|
|
Leasehold improvements
|
3-5 years
|
straight line
|
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
d) Research and Development
Research and development costs are expensed as incurred. Research and development expenses consist primarily of third party service costs under research and development agreements, salaries and related personnel costs, including stock-based compensation, costs to acquire pharmaceutical products and product rights for development and amounts paid to contract research organizations, hospitals and laboratories for the provision of services and materials for drug development and clinical trials.
Costs incurred in obtaining the licensing rights to technology in the research and development stage that have no alternative future uses and are for unapproved product compounds are expensed as incurred.
e) Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. These estimates are based on management’s best knowledge of current events and actions the Company may undertake in the future. Actual results may ultimately differ from these estimates. These estimates are reviewed periodically and as adjustments become necessary, they are reported in earnings in the period in which they become available.
f) Fair Value of Financial Instruments
The carrying amounts reported in the accompanying financial statements for cash and cash equivalents and accounts payable and accrued expenses approximate fair value because of the short‑term maturity of these financial instruments. The fair value of warrant liabilities is discussed in Note 12, and the fair value of marketable securities and certain other assets and liabilities is discussed in Note 16.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
g) Income Taxes
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. Deferred tax assets and liabilities are recorded for differences between the financial statement and tax basis of the assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates. ASC 740 requires that a valuation allowance be established when it is more likely than not that all portions of a deferred tax asset will not be realized. A review of all positive and negative evidence needs to be considered, including a company’s current and past performance, the market environment in which the company operates, length of carryback and carryforward periods and existing contracts that will result in future profits. Income tax expense is recorded for the amount of income tax payable or refundable for the period, increased or decreased by the change in deferred tax assets and liabilities during the period.
As a result of the Company’s significant cumulative losses, the Company determined that it was appropriate to establish a valuation allowance for the full amount of net deferred tax assets.
The calculation of the Company’s tax liabilities involves the inherent uncertainty associated with the application of complex tax laws. The Company is subject to examination by various taxing authorities. The Company believes that, as a result of its loss carryforward sustained to date, any examination would result in a reduction of its net operating losses rather than a tax liability. As such, the Company has not provided for any additional taxes that would be estimated under ASC 740.
h) Stock-Based Compensation
In accordance with ASC 718, “Stock Compensation,” compensation costs related to share-based payment transactions, including employee stock options, are to be recognized in the financial statements. In addition, the Company adheres to the guidance set forth within U.S. Securities and Exchange Commission (“SEC”) Staff Accounting Bulletin (“SAB”) No. 107, which provides the Staff’s views regarding the interaction between ASC 718 and certain SEC rules and regulations, and provides interpretations with respect to the valuation of share-based payments for public companies.
i) Concentration of Credit Risk
ASC 825, “Financial Instruments,” requires disclosure of any significant off balance sheet risk and credit risk concentration. The Company does not have significant off‑balance sheet risk or credit concentration. The Company maintains cash and cash equivalents with major financial institutions. From time to time the Company has funds on deposit with commercial banks that exceed federally insured limits. The balances are insured by the Federal Deposit Insurance Corporation up to $250,000. At December 31, 2017, the Company’s uninsured cash balance was $8,399,154. Management does not consider this to be a significant credit risk as the banks are large, established financial institutions.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
j) Recent Accounting Pronouncements Affecting the Company
Revenue from Contracts with Customers
In May 2014, the FASB issued Accounting Standards Update (“ASU”) 2014-09, “Revenue from Contracts with Customers,” a comprehensive new revenue recognition standard that will supersede nearly all existing revenue recognition guidance under U.S. generally accepted accounting principles. The standard’s core principle is that a company should recognize revenue when it transfers goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods and services, and provides a revenue recognition framework in accordance with this principle. On August 12, 2015, the FASB issued ASU 2015-14, which defers the effective date of ASU 2014-09 by one year to December 15, 2017 for annual reporting periods beginning after that date and interim periods therein. The Company will adopt this guidance for the annual reporting period beginning January 1, 2018, using the modified retrospective method. As the Company does not have revenue contracts, we anticipate the adoption of this guidance will not have a material impact on the operating results of the Company, there will be no significant changes to disclosures, and there will be no cumulative adjustment to the opening balance of retained earnings as of January 1, 2018.
Leases
In February 2016, the FASB issued ASU 2016-02, “Leases,” which requires an entity to recognize assets and liabilities arising from leases on the balance sheet and to provide additional disclosures about leasing arrangements. ASU 2016-02 will be effective for reporting periods beginning after December 15, 2018, with early adoption permitted. The Company is currently evaluating the impact the adoption of this guidance will have on its financial statements.
Compensation-Stock Compensation
In March 2016, the FASB issued ASU 2016-09, “Compensation-Stock Compensation: Improvements to Employee Share Based Payment Accounting,” which includes multiple provisions intended to simplify various aspects of accounting for share-based payments. The guidance is effective for reporting periods beginning after December 15, 2016, with early adoption permitted. The Company adopted this guidance during the year ended December 31, 2017. This pronouncement did not have a material impact on the financial statements.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
|
3.
|
Marketable Securities
|
The following table shows the Company’s marketable securities’ adjusted cost, gross unrealized gains and losses, and fair value by significant investment category as of December 31, 2017 and 2016:
|
December 31, 2017
|
||||||||||||||||
|
Cost
Basis
|
Gross
Unrealized
Gains
|
Gross
Unrealized
Losses
|
Fair
Value
|
|||||||||||||
|
Commercial Paper
|
$
|
3,241,005
|
$
|
-
|
$
|
(2,505
|
)
|
$
|
3,238,500
|
|||||||
|
Corporate Bonds
|
14,747,822
|
-
|
(54,381
|
)
|
14,693,441
|
|||||||||||
|
Total Marketable Securities
|
$
|
17,988,827
|
$
|
-
|
$
|
(56,886
|
)
|
$
|
17,931,941
|
|||||||
|
December 31, 2016
|
||||||||||||||||
|
Cost
Basis
|
Gross
Unrealized
Gains
|
Gross
Unrealized
Losses
|
Fair
Value
|
|||||||||||||
|
Certificates of Deposit
|
$
|
720,000
|
$
|
197
|
$
|
-
|
$
|
720,197
|
||||||||
|
Commercial Paper
|
3,987,424
|
-
|
(1,684
|
)
|
3,985,740
|
|||||||||||
|
Corporate Bonds
|
4,035,805
|
-
|
(4,635
|
)
|
4,031,170
|
|||||||||||
|
Total Marketable Securities
|
$
|
8,743,229
|
$
|
197
|
$
|
(6,319
|
)
|
$
|
8,737,107
|
|||||||
The Company typically invests in highly-rated securities, with the primary objective of minimizing the potential risk of principal loss. As of December 31, 2017, the Company had three investments of commercial paper with a fair value of $3,238,500 and unrealized losses of $2,505, and 15 corporate bonds with a fair value of $14,693,441 and unrealized losses of $54,381, all of which have been unrealized losses for less than 12 months. The Company does not intend to sell its marketable securities in an unrealized loss position. Based upon the Company’s securities’ fair value relative to the cost, high ratings, and volatility of fair value, the Company considers the declines in market value of its marketable securities to be temporary in nature and does not consider any of its investments other-than-temporarily impaired, and anticipates that it will recover the entire amortized cost basis.
The amortized cost basis and fair value of marketable securities by contractual maturity are:
|
Maturity
|
Cost Basis
|
Fair Value
|
||||||
|
Less than 1 year
|
$
|
11,981,457
|
$
|
11,955,101
|
||||
|
1 to 5 years
|
6,007,370
|
5,976,840
|
||||||
|
Total Marketable Securities
|
$
|
17,988,827
|
$
|
17,931,941
|
||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 4. |
Prepaid Expenses and Other Current Assets
|
|
December 31,
2017
|
December 31,
2016
|
|||||||
|
Deposits on contracts
|
$
|
793,940
|
$
|
179,476
|
||||
|
Prepaid expenses and other current assets
|
510,601
|
429,041
|
||||||
|
$
|
1,304,541
|
$
|
608,517
|
|||||
Deposits on contracts consist of deposits on research and development contracts for services that had not been incurred as of the balance sheet date. Prepaid expenses and other assets include prepaid general and administrative expenses, such as insurance, rent, investor relations fees and compensatory stock issued for services not yet incurred as of the balance sheet date.
| 5. |
Equipment, Net
|
|
December 31,
2017
|
December 31,
2016
|
|||||||
|
Furniture and fixtures
|
$
|
82,686
|
$
|
78,794
|
||||
|
Office and computer equipment
|
171,724
|
113,932
|
||||||
|
Lab equipment
|
445,134
|
431,650
|
||||||
|
Leasehold improvements
|
133,762
|
133,762
|
||||||
|
Total equipment
|
833,306
|
758,138
|
||||||
|
Less: Accumulated depreciation and amortization
|
(711,846
|
)
|
(669,488
|
)
|
||||
|
Net carrying amount
|
$
|
121,460
|
$
|
88,650
|
||||
| 6. |
Accounts Payable and Accrued Expenses
|
|
December 31,
2017
|
December 31,
2016
|
|||||||
|
Trade payables
|
$
|
895,638
|
$
|
430,013
|
||||
|
Accrued expenses
|
95,416
|
141,190
|
||||||
|
Accrued research and development contract costs
|
1,435,109
|
499,889
|
||||||
|
Payroll liabilities
|
807,763
|
811,408
|
||||||
|
$
|
3,233,926
|
$
|
1,882,500
|
|||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 7. |
Deferred Research and Development Arrangements
|
Rexgene Biotech Co., Ltd.
In 2003, the Company entered into a collaborative research agreement with Rexgene Biotech Co., Ltd. (“Rexgene”), a stockholder, pursuant to which Rexgene agreed to assist the Company with the research, development and clinical trials necessary for registration of the Company’s drug candidate RX-0201 (Archexin®) in Asia. In accordance with the agreement, Rexgene paid the Company a one-time fee of $1,500,000 in 2003. The agreement provided that it would expire upon the later of (i) 20 years after the date of the agreement or (ii) the expiration of the patents relating to RX-0201. The amortization reduces research and development expenses for the periods presented.
The Company is using 20 years as its basis for recognition and accordingly research and development expenses were reduced by $75,000 for each of the years ended December 31, 2017, 2016 and 2015. The remaining $375,000 and $450,000 to be amortized at December 31, 2017 and 2016, respectively, are reflected as a deferred research and development arrangement on the balance sheet. The payment from Rexgene is being used in the cooperative funding of the costs of development of RX-0201.
On February 5, 2018, the Company and NEXT BT Co. Ltd., the successor in interest to Rexgene, terminated the agreement.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 8. |
Other Liabilities
|
Deferred Lease Incentive
In accordance with the Company’s office lease agreement, as amended and further discussed in Note 15, the Company has been granted leasehold improvement allowances from the lessor to be used for the construction cost of improvements to the leased property, which included architectural and engineering fees, government agency plan check, permit and other fees, sales and use taxes, testing and inspection costs and telephone and data cabling and wiring in the premises. The Company accounted for the benefit of the leasehold improvement allowance as a reduction of rental expense over the term of the office lease.
The following table sets forth the cumulative deferred lease incentive:
|
December 31,
2017
|
December 31,
2016
|
|||||||
|
Deferred lease incentive
|
$
|
154,660
|
$
|
154,660
|
||||
|
Less accumulated amortization
|
(135,995
|
)
|
(123,551
|
)
|
||||
|
Balance
|
$
|
18,665
|
$
|
31,109
|
||||
Deferred Office Lease Expense
The lease agreement, as amended, provided for an initial annual base rent with annual increases over the lease term. The Company recognizes rental expense on a straight-line basis over the term of the lease, which resulted in a deferred rent liability of $38,059 and $48,095 as of December 31, 2017 and 2016, respectively.
| 9. |
Net Loss per Common Share
|
Basic loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding for the period. Diluted loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding, plus the number of common share equivalents that would be dilutive. As of December 31, 2017, 2016 and 2015, there were stock options, restricted stock units and warrants to acquire, in the aggregate, 8,961,140, 7,142,728, and 3,908,295 shares of the Company’s common stock, respectively, which are potentially dilutive. However, diluted loss per share for all periods presented is the same as basic loss per share because the inclusion of common share equivalents would be anti-dilutive.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 10. |
Common Stock
|
The following transactions occurred during the years ended December 31, 2017, 2016 and 2015:
Reverse Stock Split
On May 5, 2017, the Company effected a one-for-ten reverse stock split of the outstanding shares of the Company’s common stock, together with a corresponding proportional reduction in the number of authorized shares of the Company’s capital stock. Each ten shares of the Company’s common stock, par value $0.0001 per share, issued and outstanding at the effective time of the reverse stock split were reclassified and combined into one share of common stock par value $0.0001 per share. The number of shares of common stock and preferred stock the Company is authorized to issue was reduced to 50 million and 10 million, respectively. All share and per share amounts of common stock, stock options, stock warrants and restricted stock units have been restated for all periods to give retroactive effect to the reverse stock split. Accordingly, an amount equal to the par value of the decreased shares resulting from the reverse stock split was reclassified from “Common stock” to “Additional paid-in capital.”
Public Offerings
November 2015
On November 12, 2015, the Company closed a registered direct public offering of 1,666,667 shares of common stock and warrants to purchase up to 1,250,000 shares of common stock. The common stock and warrants were sold in units, consisting of a share of common stock and a warrant to purchase 0.75 shares of common stock, at a price of $4.20 per unit, with an exercise price for the warrants of $5.30 per share. The total gross proceeds of the offering were $7,000,000. The warrants issued became exercisable beginning six months after the closing date, and will remain exercisable until the five-year anniversary of the initial exercise date, and were recorded as liabilities at fair value.
A summary of the allocation of the proceeds of the offering is shown below:
|
Gross Proceeds:
|
$
|
7,000,000
|
||
|
Allocated to warrant liabilities:
|
2,792,500
|
|||
|
Allocated to common stock and additional paid-in capital
|
4,207,500
|
|||
|
Total allocated gross proceeds:
|
$
|
7,000,000
|
The closing costs of $740,323 included 83,333 warrants valued at $174,417 and $565,906 for placement agent and other fees. Based upon the estimated fair value of the stock and warrants in the units, the Company allocated $211,116 to financing expense and $529,207 as stock issuance costs.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
March 2016
On March 2, 2016, the Company closed a registered direct public offering of 1,562,500 shares of common stock and warrants to purchase up to 1,171,875 shares of common stock. The common stock and warrants were sold in units, consisting of a share of common stock and a warrant to purchase 0.75 shares of common stock, at a price of $3.20 per unit, with an exercise price for the warrants of $4.20 per share. The total gross proceeds of the offering were $5,000,000. The issued warrants issued became exercisable beginning six months after the closing date, and will remain exercisable until the five-year anniversary of the initial exercise date, and were recorded as liabilities at fair value.
A summary of the allocation of the proceeds of the offering is shown below:
|
Gross Proceeds:
|
$
|
5,000,000
|
||
|
Allocated to warrant liabilities:
|
2,419,922
|
|||
|
Allocated to common stock and additional paid-in capital
|
2,580,078
|
|||
|
Total allocated gross proceeds:
|
$
|
5,000,000
|
The closing costs of $575,751 included 78,125 warrants valued at $155,938 and $419,813 for placement agent and other fees. Based upon the estimated fair value of the stock and warrants in the units, the Company allocated $169,887 to financing expense and $405,864 as stock issuance costs.
September 2016
On September 19, 2016, the Company closed a registered direct public offering of 2,400,000 shares of common stock and warrants to purchase up to 1,800,000 shares of common stock. The common stock and warrants were sold in units, consisting of a share of common stock and a warrant to purchase 0.75 shares of common stock, at a price of $2.50 per unit, with an exercise price for the warrants of $3.00 per share. The total gross proceeds of the offering were $6,000,000. The warrants issued became exercisable beginning six months after the closing date, and will remain exercisable until the five-year anniversary of the initial exercise date, and were recorded as liabilities at fair value.
A summary of the allocation of the proceeds of the offering is shown below:
|
Gross Proceeds:
|
$
|
6,000,000
|
||
|
Allocated to warrant liabilities:
|
1,671,120
|
|||
|
Allocated to common stock and additional paid-in capital
|
4,328,880
|
|||
|
Total allocated gross proceeds:
|
$
|
6,000,000
|
The closing costs of $575,094 included 144,000 warrants valued at $117,130 and $457,964 for placement agent and other fees. Based upon the estimated fair value of the stock and warrants in the units, the Company allocated $143,203 to financing expense and $431,891 as stock issuance costs.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
June 2017
On June 12, 2017 the Company closed a registered direct public offering of 3,030,304 shares of common stock and warrants to purchase up to 1,515,152 shares of common stock. The common stock and warrants were sold in units, consisting of a share of common stock and a warrant to purchase 0.5 shares of common stock, at a price of $3.30 per unit, with an exercise price for the warrants of $4.00 per share. The total gross proceeds of the offering were $10,000,003. The warrants issued became exercisable beginning six months after the closing date, and will remain exercisable until the five-year anniversary of the initial exercise date, and were recorded as liabilities at fair value.
A summary of the allocation of the proceeds of the offering is shown below:
|
Gross Proceeds:
|
$
|
10,000,003
|
||
|
Allocated to warrant liabilities
|
3,673,168
|
|||
|
Allocated to common stock and additional paid-in capital
|
6,326,835
|
|||
|
Total allocated gross proceeds:
|
$
|
10,000,003
|
The Company also issued warrants to purchase up to an aggregate 181,818 shares of common stock to the placement agent in the offering. The closing costs for the offering of $1,193,052 included $434,320 for the placement agent warrants and $758,732 for placement agent and other fees. Based on the estimated fair value of the stock and warrants in the units, the Company allocated $333,050 to financing expense for the warrants and $860,002 as stock issuance costs.
October 2017
On October 17, 2017 the Company closed a registered direct public offering of 3,265,309 shares of common stock and warrants to purchase up to 1,632,654 shares of common stock. The common stock and warrants were sold in units, consisting of a share of common stock and a warrant to purchase 0.5 shares of common stock, at a price of $2.45 per unit, with an exercise price for the warrants of $2.85 per share. The total gross proceeds of the offering were $8,000,007. The warrants issued will become exercisable beginning six months after the closing date, and will remain exercisable until the five-year anniversary of the initial exercise date, and were recorded as liabilities at fair value.
A summary of the allocation of the proceeds of the offering is shown below:
|
Gross Proceeds:
|
$
|
8,000,007
|
||
|
Allocated to warrant liabilities
|
2,360,459
|
|||
|
Allocated to common stock and additional paid-in capital
|
5,639,548
|
|||
|
Total allocated gross proceeds:
|
$
|
8,000,007
|
The Company also issued warrants to purchase up to an aggregate 195,919 shares of common stock to the placement agent in the offering. The closing costs for the offering of $830,111 included $270,754 for the placement agent warrants and $559,357 for placement agent and other fees. Based on the estimated fair value of the stock and warrants in the units, the Company allocated $219,577 to financing expense for the warrants and $610,534 as stock issuance costs.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
At Market Offering
On August 2, 2017, the Company terminated the at market issuance sales agreement (the “Sales Agreement”), dated March 16, 2015, with MLV & Co. LLC, now part of FBR & Co. (“MLV”), pursuant to which the Company was entitled to issue and sell shares of its common stock having an aggregate offering price of up to $40 million from time to time, at its option, through MLV as the Company’s sales agent. For the year ended December 31, 2015, the Company sold 140,707 shares of common stock pursuant to the Sales Agreement for $1,042,573 in gross proceeds at a weighted average price of $7.41 per share. Net proceeds to the Company were $1,005,715 after deducting commissions and other transaction costs. There were no shares sold under the Sales Agreement for the years ended December 31, 2017 and 2016.
Compensatory Shares
The Company has issued restricted shares to vendors in exchange for services. The table below summarizes the shares issued and the related market value:
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Compensatory shares issued
|
15,000
|
33,000
|
15,000
|
|||||||||
|
Aggregate market value
|
$
|
31,200
|
$
|
97,649
|
$
|
102,000
|
||||||
Stock Option and Stock Warrant Exercises
The table below summarizes stock options and stock warrants exercised:
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Stock Option Exercises
|
||||||||||||
|
Number of shares issued
|
25,000
|
-
|
88,943
|
|||||||||
|
Total cash received
|
$
|
77,500
|
$
|
-
|
$
|
708,617
|
||||||
|
Stock Warrant Exercises
|
||||||||||||
|
Number of shares issued
|
1,652,623
|
-
|
4,730
|
|||||||||
|
Total cash received
|
$
|
5,354,093
|
$
|
-
|
$
|
22,325
|
||||||
Treasury Stock Transactions
On December 3, 2015, the Company retired 11,321 shares of treasury stock with an aggregate purchase price of $128,410.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 11. |
Stock-Based Compensation
|
As of December 31, 2017, the Company had 1,814,231 options to purchase common stock and 47,300 restricted stock units (“RSUs”) outstanding.
At the Company’s Annual Meeting of the Stockholders held on June 10, 2013, the Company’s stockholders voted to approve the Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan (the “2013 Plan”). Under the 2013 Plan, the Company grants equity awards to key employees, directors and consultants of the Company. At the Company’s Annual Meeting held on June 9, 2016, the Company’s stockholders voted to approve an amendment and restatement of the 2013 Plan, including to provide for awards of restricted stock and restricted stock units. The Company initially reserved 1,700,000 shares of common stock for issuance pursuant to the 2013 Plan, and on April 11, 2017, the Company’s stockholders approved an increase of 1,700,000 shares of common stock reserved for issuance pursuant to the 2013 Plan. As of December 31, 2017, there were 1,477,231 options and 47,300 RSUs outstanding under the 2013 Plan, and 1,874,719 shares were available for issuance.
On August 5, 2003, the Company established a stock option plan (the “2003 Plan”). Under the 2003 Plan, the Company granted stock options to key employees, directors and consultants of the Company. With the adoption of the 2013 Plan, no new stock options may be issued under the 2003 Plan, but previously issued options under the 2003 Plan remain outstanding until their expiration. As of December 31, 2017, there were 325,000 outstanding options under the 2003 Plan.
In March 2016, the Company granted to a third party an option to purchase up to 12,000 shares of the Company’s common stock. Of the Company’s outstanding options as of December 31, 2017, these were the only options that were not issued pursuant to the 2013 Plan or the 2003 Plan.
Accounting for Awards
Stock-based compensation expense is the estimated fair value of options and RSUs granted amortized on a straight-line basis over the requisite vesting service period for the entire portion of the award. Total stock-based compensation recognized by the Company for the years ended December 31, 2017, 2016 and 2015 is as follows:
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Statement of operations line item:
|
||||||||||||
|
General and administrative
|
$
|
765,726
|
$
|
905,911
|
$
|
665,063
|
||||||
|
Research and development
|
278,441
|
503,577
|
372,616
|
|||||||||
|
Total
|
$
|
1,044,167
|
$
|
1,409,488
|
$
|
1,037,679
|
||||||
No income tax benefit has been recognized in the statement of operations for stock-based compensation arrangements as the Company has provided for a 100% valuation allowance on its net deferred tax assets.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
Summary of Stock Option Transactions
There were 483,260 stock options granted at exercise prices ranging from $1.84 to $6.18 with an aggregate fair value of $738,937 during the year ended December 31, 2017. There were 592,637 stock options granted at exercise prices ranging from $1.80 to $3.70 with an aggregate fair value of $1,156,273 during the year ended December 31, 2016. There were 420,130 stock options granted at exercise prices ranging from $5.40 to $8.90 with an aggregate fair value of $1,994,893 during the year ended December 31, 2015.
For the majority of the grants to employees, the vesting period is either (i) 30%, 30% and 40% on the first, second and third anniversaries, of the grant date, respectively, or (ii) 25% each on the first four anniversaries. Options expire between five and ten years from the date of grant. For grants to non-employee consultants of the Company, the vesting period is between one and three years, subject to the fulfillment of certain conditions in the individual stock agreements, or 100% upon the occurrence of certain events specified in the individual stock agreements.
The fair value of options at the date of grant was estimated using the Black-Scholes option pricing model. The Company took into consideration guidance under ASC 718, “Compensation-Stock Compensation” and Staff Accounting Bulletin No. 107 (“SAB 107”) when reviewing and updating assumptions.
Significant assumptions are determined as follows:
Expected Term. The expected term is estimated using the simplified method whereby the expected term equals the arithmetic average of the vesting term and the original contractual term of the option.
Volatility. Volatility is based on the historical trading volatility of the Company’s stock on the date of grant for a period consistent with the expected term.
Risk-Free Interest Rate. The risk-free interest rate is based on the zero-coupon U.S. Treasury instruments on the date of grant with a maturity date consistent with the expected term of the Company’s stock option grants.
Expected Dividend. To date, the Company has not declared or paid any cash dividends and do not have any plans to do so in the future. Therefore, the Company used an expected dividend yield of zero.
The assumptions made in calculating the fair values of options are as follows:
|
Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Black-Scholes assumptions
|
||||||||||||
|
Expected dividend yield
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||
|
Expected volatility
|
69-79
|
%
|
31-75
|
%
|
72-80
|
%
|
||||||
|
Risk-free interest rate
|
1.7-2.0
|
%
|
0.8-1.4
|
%
|
1.2-1.7
|
%
|
||||||
|
Expected term (in years)
|
5.5-6 years
|
2-6 years
|
5-6 years
|
|||||||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
The following table summarizes share-based transactions:
|
Number of
Options
|
Weighted
Average
Exercise
Price
|
Weighted Average
Remaining
Contractual Term
|
Aggregate
Intrinsic
Value
|
||||||||||
|
Outstanding, January 1, 2017
|
1,690,037
|
$
|
6.20
|
7.3 years
|
$
|
-
|
|||||||
|
Granted
|
483,260
|
$
|
2.37
|
||||||||||
|
Exercised
|
(25,000
|
)
|
$
|
3.10
|
|||||||||
|
Expired
|
(20,000
|
)
|
$
|
16.18
|
|||||||||
|
Cancelled
|
(314,066
|
)
|
$
|
4.97
|
|||||||||
|
Outstanding, December 31, 2017
|
1,814,231
|
$
|
5.33
|
7.1 years
|
$
|
53,883
|
|||||||
|
Exercisable, December 31, 2017
|
1,086,688
|
$
|
6.50
|
6.2 years
|
$
|
321
|
|||||||
The total intrinsic value of options exercised was $97,872 and $99,895 for the years ended December 31, 2017 and 2015, respectively. There were no stock options exercised during the year ended December 31, 2016. The weighted average fair value of options granted was $1.53, $2.00, and $4.70 for the years ended December 31, 2017, 2016 and 2015, respectively.
A summary of the Company’s unvested options as of December 31, 2017 and changes during the year ended December 31, 2017 is presented below:
|
2017
|
||||||||
|
Number of Options
|
Weighted Average Fair
Value at Grant Date
|
|||||||
|
Unvested at January 1, 2017
|
897,123
|
$
|
3.21
|
|||||
|
Granted
|
483,260
|
$
|
1.53
|
|||||
|
Vested
|
(489,235
|
)
|
$
|
3.11
|
||||
|
Cancelled
|
(163,605
|
)
|
$
|
2.25
|
||||
|
Unvested at December 31, 2017
|
727,543
|
$
|
2.39
|
|||||
As of December 31, 2017, there was $1,233,528 of total unrecognized compensation cost related to unvested stock options, which is expected to be recognized over a weighted average vesting period of 2.3 years.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
Summary of Restricted Stock Unit Transactions
The Company began granting RSUs to employees in 2017. There were 62,300 RSUs granted with an aggregate fair value of $114,632 during the year ended December 31, 2017. The fair value of an RSU award is the closing price of the Company’s common stock on the date of grant.
A summary of RSU activity for the year ended December 31, 2017 is as follows:
|
Number of RSUs
|
Weighted
Average Grant
Date Fair Value
|
|||||||
|
Outstanding, January 1, 2017
|
-
|
$
|
-
|
|||||
|
Granted
|
62,300
|
$
|
1.84
|
|||||
|
Vested and Released
|
-
|
$
|
-
|
|||||
|
Cancelled
|
(15,000
|
)
|
$
|
1.84
|
||||
|
Outstanding, December 31, 2017
|
47,300
|
$
|
1.84
|
|||||
As of December 31, 2017, there was $67,496 of total unrecognized compensation cost related to unvested RSUs which is expected to be recognized over a weighted average vesting period of 3.2 years.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
|
12.
|
Warrants
|
As of December 31, 2017, warrants to purchase 7,099,609 shares were outstanding, having exercise prices ranging from $2.85 to $12.80 and expiration dates ranging from July 26, 2018 to April 17, 2023.
|
2017
|
2016
|
|||||||||||||||
|
Number of
warrants
|
Weighted
average exercise
price
|
Number of
warrants
|
Weighted average
exercise price
|
|||||||||||||
|
Balance, January 1
|
5,452,691
|
$
|
4.92
|
2,649,199
|
$
|
7.97
|
||||||||||
|
Issued during the period
|
3,525,543
|
$
|
3.42
|
3,194,000
|
$
|
3.47
|
||||||||||
|
Exercised during the period
|
(1,861,195
|
)
|
$
|
3.51
|
-
|
$
|
-
|
|||||||||
|
Expired during the period
|
(17,430
|
)
|
$
|
4.72
|
(390,508
|
)
|
$
|
13.72
|
||||||||
|
Balance, December 31
|
7,099,609
|
$
|
4.55
|
5,452,691
|
$
|
4.92
|
||||||||||
At December 31, 2017, the weighted average remaining contractual life of the outstanding warrants was 4.0 years.
The warrants issued to investors in the December 2012, November 2015, March 2016 and September 2016 offerings contain a provision for net cash settlement in the event of a fundamental transaction (contractually defined to include a merger, sale of substantially all assets, tender offer or share exchange). Pursuant to the November 2015, March 2016, and September 2016 warrants, if fundamental transaction occurs, then the warrant holder has the option to receive cash, equal to the fair value of the remaining unexercised portion of the warrant. The option is available to holders of the December 2012 warrants only if the consideration issued in the fundamental transaction consists of cash or stock in a non-public company. The June 2017 and October 2017 warrants contain a provision that allows the holder to opt for cash settlement in a fundamental transaction that was approved by, or required to be approved by, the board of directors of the Company. All of the Company’s outstanding warrants provide the holder the option as to the type of consideration received if the holders of common stock receive an option as to their consideration. In addition, all of the Company’s outstanding warrants contain a cashless exercise provision that is exercisable only in the event that a registration statement is not effective. That provision may not be operative if an effective registration statement is not available because an exemption under the U.S. securities laws may not be available to issue unregistered shares. As a result, net cash settlement may be required, and the warrants require liability classification.
ASC 820 provides requirements for disclosure of liabilities that are measured at fair value on a recurring basis in periods subsequent to the initial recognition. Fair values for warrants were determined using the Binomial Lattice (“Lattice”) valuation technique. The Lattice model provides for dynamic assumptions regarding volatility and risk-free interest rates within the total period to maturity. Accordingly, within the contractual term, the Company provided multiple date intervals over which multiple volatilities and risk-free interest rates were used. These intervals allow the Lattice model to project outcomes along specific paths that consider volatilities and risk-free rates that would be more likely in an early exercise scenario.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
Significant assumptions are determined as follows:
Trading market values—Published trading market values;
Exercise price—Stated exercise price;
Term—Remaining contractual term of the warrant;
Volatility—Historical trading volatility for periods consistent with the remaining terms; and
Risk-free rate—Yields on zero coupon government securities with remaining terms consistent with the remaining terms of the warrants.
Due to the fundamental transaction provision, which could provide for early redemption of the warrants, the model also considered the probability the Company would enter into a fundamental transaction during the remaining term of the warrant. Because the Company is not yet achieving positive cash flow, management believes the probability of a fundamental transaction occurring over the term of the warrant is unlikely and therefore estimates the probability of entering into a fundamental transaction to be 5%. For valuation purposes, the Company also assumed that if such a transaction did occur, it was more likely to occur towards the end of the term of the warrants.
The significant unobservable inputs used in the fair value measurement of the warrants include management’s estimate of the probability that a fundamental transaction may occur in the future. Significant increases (decreases) in the probability of occurrence would result in a significantly higher (lower) fair value measurement.
The following table summarizes the fair value of the warrants as of the respective balance sheet dates:
|
Fair Value as of:
|
||||||||
|
Warrant Issuance:
|
December 31, 2017
|
December 31, 2016
|
||||||
|
December 2012 Investor Warrants
|
$
|
-
|
$
|
49
|
||||
|
July 2013 Investor Warrants
|
8,762
|
2,060
|
||||||
|
October 2013 Investor Warrants
|
26,288
|
3,708
|
||||||
|
January 2014 Investor Warrants
|
29,257
|
714
|
||||||
|
November 2015 Investor Warrants
|
1,260,050
|
260,500
|
||||||
|
November 2015 Placement Agent Warrants
|
2,936
|
13,542
|
||||||
|
March 2016 Investor Warrants
|
697,554
|
358,945
|
||||||
|
March 2016 Placement Agent Warrants
|
-
|
21,320
|
||||||
|
September 2016 Investor Warrants
|
1,054,083
|
854,640
|
||||||
|
September 2016 Placement Agent Warrants
|
-
|
57,888
|
||||||
|
June 2017 Investor Warrants
|
1,981,864
|
-
|
||||||
|
June 2017 Placement Agent Warrants
|
221,591
|
-
|
||||||
|
October 2017 Investor Warrants
|
2,305,552
|
-
|
||||||
|
October 2017 Placement Agent Warrants
|
265,698
|
-
|
||||||
|
Total:
|
$
|
7,853,635
|
$
|
1,573,366
|
||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
The following table summarizes the number of shares indexed to the warrants as of the respective balance sheet dates:
|
Number of Shares indexed as of:
|
||||||||
|
Warrant Issuance
|
December 31, 2017
|
December 31, 2016
|
||||||
|
December 2012 Investor Warrants
|
-
|
17,430
|
||||||
|
July 2013 Investor Warrants
|
200,000
|
200,000
|
||||||
|
October 2013 Investor Warrants
|
231,732
|
231,732
|
||||||
|
January 2014 Investor Warrants
|
476,193
|
476,193
|
||||||
|
November 2015 Investor Warrants
|
1,250,001
|
1,250,001
|
||||||
|
November 2015 Placement Agent Warrants
|
3,334
|
83,335
|
||||||
|
March 2016 Investor Warrants
|
607,806
|
1,171,875
|
||||||
|
March 2016 Placement Agent Warrants
|
-
|
78,125
|
||||||
|
September 2016 Investor Warrants
|
805,000
|
1,800,000
|
||||||
|
September 2016 Placement Agent Warrants
|
-
|
144,000
|
||||||
|
June 2017 Investor Warrants
|
1,515,152
|
-
|
||||||
|
June 2017 Placement Agent Warrants
|
181,818
|
-
|
||||||
|
October 2017 Investor Warrants
|
1,632,654
|
-
|
||||||
|
October 2017 Placement Agent Warrants
|
195,919
|
-
|
||||||
|
Total:
|
7,099,609
|
5,452,691
|
||||||
The assumptions used in calculating the fair values of the warrants are as follows:
|
December 31, 2017
|
December 31, 2016
|
|||||||
|
Trading market prices
|
$
|
2.02
|
$
|
1.40
|
||||
|
Estimated future volatility
|
104
|
%
|
104
|
%
|
||||
|
Dividend
|
-
|
-
|
||||||
|
Estimated future risk-free rate
|
2.14-2.45
|
%
|
1.06-2.44
|
%
|
||||
|
Equivalent volatility
|
85-104
|
%
|
51-60
|
%
|
||||
|
Equivalent risk-free rate
|
1.30-1.89
|
%
|
0.59-1.25
|
%
|
||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
Changes in the fair value of the warrant liabilities, carried at fair value, as reported as “unrealized (loss) gain on fair value of warrants” in the statement of operations:
|
For the Year Ended December 31,
|
||||||||||||
|
2017
|
2016
|
2015
|
||||||||||
|
Expired Warrants
|
$
|
-
|
$
|
2,590
|
$
|
458,439
|
||||||
|
December 2012 Investor Warrants
|
49
|
9,769
|
70,856
|
|||||||||
|
July 2013 Investor Warrants
|
(6,702
|
)
|
119,360
|
666,894
|
||||||||
|
October 2013 Investor Warrants
|
(22,580
|
)
|
165,641
|
780,407
|
||||||||
|
January 2014 Investor Warrants
|
(28,543
|
)
|
130,762
|
1,347,724
|
||||||||
|
November 2015 Investor Warrants
|
(999,550
|
)
|
1,908,875
|
623,125
|
||||||||
|
November 2015 Placement Agent Warrants
|
(365,748
|
)
|
121,593
|
39,282
|
||||||||
|
March 2016 Investor Warrants
|
(2,708,163
|
)
|
2,060,977
|
-
|
||||||||
|
March 2016 Placement Agent Warrants
|
(351,899
|
)
|
134,617
|
-
|
||||||||
|
September 2016 Investor Warrants
|
(4,571,872
|
)
|
816,480
|
-
|
||||||||
|
September 2016 Placement Agent Warrants
|
(503,150
|
)
|
59,243
|
-
|
||||||||
|
June 2017 Investor Warrants
|
1,691,304
|
-
|
-
|
|||||||||
|
June 2017 Placement Agent Warrants
|
212,729
|
-
|
-
|
|||||||||
|
October 2017 Investor Warrants
|
54,907
|
-
|
-
|
|||||||||
|
October 2017 Placement Agent Warrants
|
5,056
|
-
|
-
|
|||||||||
|
Total:
|
$
|
(7,594,162
|
)
|
$
|
5,529,907
|
$
|
3,986,727
|
|||||
| 13. |
Mediation Settlement
|
In connection with the process of seeking patent protection for RX-5902 in Japan, the Company had filed a patent application including claims covering RX-5902 with the Japanese Patent Office (“JPO”) for examination. The JPO initially agreed that the claims covering the compound for RX-5902 were allowable, but as a result of a mistake in the patent application filing as prepared and submitted by the Company’s Japanese patent attorney and incomplete review by the JPO’s patent examiner, the JPO issued a decision to grant a patent with claims that did not include RX-5902’s chemical structure. The Company appealed this decision with the JPO to request withdrawal of the decision to grant so that the correct claims would be allowed, but the JPO refused to withdraw its decision. As a result, and in accordance with Japanese law and procedure for appealing patent application decisions, the Company has filed a lawsuit against the JPO in Tokyo District Court to cause the JPO to reverse its decision to grant the errant patent and to allow a patent that includes claims covering RX-5902. The patent application at issue remains pending subject to the outcome of this action. While the composition of matter patent on RX-5902 structure remains pending in Japan, the Company either has already or will have protection in Japan from its issued and pending patents on formulation, method of use, and method of manufacturing as well as from market exclusivity period.
On December 19, 2016, the Company entered into a binding settlement arrangement with the Company’s Japanese patent attorney in which the Japanese patent attorney agreed to pay a one-time settlement JPY 210,000,000, or $1,770,658, in exchange for the Company agreeing not to bring any future claims on account of this patent filing. The settlement payment was received by the Company by December 31, 2016.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 14. |
Income Taxes
|
No provision for federal and state income taxes was required for the years ended December 31, 2017, 2016 and 2015 due to the Company’s operating losses and increased deferred tax asset valuation allowance. At December 31, 2017 and 2016, the Company had unused net operating loss carry-forwards of approximately $127,877,000 and $111,605,000, respectively, which expire at various dates through 2037. Some of this amount may be subject to annual limitations under certain provisions of the Internal Revenue Code related to “changes in ownership.”
As of December 31, 2017, and 2016, the deferred tax assets related to the aforementioned carry-forwards have been fully offset by valuation allowances, because significant utilization of such amounts is not presently expected in the foreseeable future.
Deferred tax assets and valuation allowances consist of:
|
December 31,
2017
|
December 31,
2016
|
|||||||
|
Net Operating Loss Carryforwards
|
$
|
35,805,000
|
$
|
43,526,000
|
||||
|
Stock Compensation Expense
|
1,458,000
|
1,968,000
|
||||||
|
Book tax differences on assets and liabilities
|
365,000
|
547,000
|
||||||
|
Valuation Allowance
|
(37,628,000
|
)
|
(46,041,000
|
)
|
||||
|
Net Deferred Tax Assets
|
$
|
-
|
$
|
-
|
||||
The Company files income tax returns in the U.S. federal and Maryland state jurisdictions. Tax years for fiscal 2014 through 2017 are open and potentially subject to examination by the federal and Maryland state taxing authorities.
The Tax Cuts and Jobs Act, which was signed into law on December 22, 2017, reduces the U.S. corporate income tax rate from 35 percent to 21 percent. The Company remeasured its net deferred tax assets based on the new corporate tax rate. There was no impact on income tax expenses resulting from the remeasurement due to the full offset by the valuation allowance.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 15. |
Commitments and Contingencies
|
| a) |
The Company has contracted with various vendors for research and development services, with terms that require payments over the term of the agreements, usually ranging from two to 36 months. The costs to be incurred are estimated and are subject to revision. As of December 31, 2017, the total estimated cost to complete these agreements was approximately $11,110,000. All of these agreements may be terminated by either party upon appropriate notice as stipulated in the respective agreements.
|
| b) |
On June 22, 2009, the Company entered into a License Agreement with Korea Research Institute of Chemical Technology (“KRICT”) to acquire the rights to all intellectual property related to quinoxaline-piperazine derivatives that were synthesized under a Joint Research Agreement. The initial license fee was $100,000, all of which was paid as of December 31, 2009. The agreement with KRICT calls for a one-time milestone payment of $1,000,000 within 30 days after the first achievement of marketing approval of the first commercial product arising out of or in connection with the use of KRICT’s intellectual properties. As of December 31, 2017, the milestone has not occurred.
|
| c) |
Office Space Lease
|
On June 5, 2009, the Company entered into a commercial lease agreement for 5,466 square feet of office space in Rockville, Maryland. The lease was amended on June 7, 2013 to extend the term until June 30, 2019.
On July 26, 2014, the lease was amended to add 1,727 square feet of office space, beginning on September 1, 2014 and ending on August 31, 2015. The lease of additional space was subsequently renewed until June 30, 2019. Under the lease agreement, the Company pays its allocable portion of real estate taxes and common area operating charges.
Rent paid under the Company’s lease during the years ended December 31, 2017, 2016, and 2015 was $206,667, $205,324, and $202,529, respectively.
Prior Laboratory Lease
On August 26, 2014, the Company signed a one-year renewal to use laboratory space commencing on July 1, 2014. The lease required monthly rental payments of $4,554. Rent paid under the Company’s lease during the year ended December 31, 2015 was $27,324.
Current Laboratory Lease
On April 20, 2015, the Company signed a five-year lease agreement for 2,552 square feet of laboratory space commencing on July 1, 2015 and ending on June 30, 2020. Under the lease agreement, the Company pays its allocable portion of real estate taxes and common area operating charges. Rent paid under this lease during the years ended December 31, 2017, 2016 and 2015 was $64,032, $62,167 and $30,624, respectively.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
Future rental payments over the next five years for all leases are as follows:
|
For the year ending December 31:
|
2018
|
$
|
279,274
|
||
|
|
2019 |
176,080
|
|||
|
2020
|
34,468
|
||||
|
|
Total
|
$
|
489,822
|
| d) |
The Company has established a 401(k) plan for its employees. The Company has elected to match 100% of the first 3% of an employee’s compensation plus 50% of an additional 2% of the employee’s deferral. Expense related to this matching contribution aggregated to $123,145, $113,204, and $121,519, for the years ended December 31, 2017, 2016 and 2015 respectively.
|
| e) |
In July 2013, the Company entered into an exclusive license agreement with the University of Maryland, Baltimore for a novel drug delivery platform, Nano-Polymer Drug Conjugate Systems. The agreement requires the Company to make payments to the University of Maryland if any products from the licensed delivery platform achieve development milestones. As of December 31, 2017, no development milestones have occurred.
|
| f) |
In October 2013, the Company signed an exclusive license agreement with the Ohio State Innovation Foundation, for a novel oligonucleotide drug delivery platform, Lipid-Coated Albumin Nanoparticle. The agreement requires the Company to make payments to the Ohio State Innovation Foundation or any products from the licensed delivery platform achieve development milestones. As of December 31, 2017, no development milestones have occurred.
|
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
|
16.
|
Fair Value Measurements
|
ASC 820 defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, not adjusted for transaction costs. ASC 820 also establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels giving the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1) and the lowest priority to unobservable inputs (Level 3).
The three levels are described below:
|
Level 1 Inputs
|
—
|
Unadjusted quoted prices in active markets for identical assets or liabilities that are accessible by the Company;
|
|
|
Level 2 Inputs
|
—
|
Quoted prices in markets that are not active or financial instruments for which all significant inputs are observable, either directly or indirectly;
|
|
|
Level 3 Inputs
|
—
|
Unobservable inputs for the asset or liability including significant assumptions of the Company and other market participants.
|
The following tables present assets and liabilities that are measured at fair value on a recurring basis and are categorized using the fair value hierarchy. There have been no changes in the methodologies used at December 31, 2017 and 2016.
|
Fair Value Measurements at December 31, 2017
|
||||||||||||||||
|
Total
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Assets:
|
||||||||||||||||
|
Commercial Paper
|
3,238,500
|
-
|
3,238,500
|
-
|
||||||||||||
|
Corporate Bonds
|
14,693,441
|
-
|
14,693,441
|
-
|
||||||||||||
|
Total Assets:
|
$
|
17,931,941
|
$
|
-
|
$
|
17,931,941
|
$
|
-
|
||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrant Liabilities
|
$
|
7,853,635
|
$
|
-
|
$
|
-
|
$
|
7,853,635
|
||||||||
|
Fair Value Measurements at December 31, 2016
|
||||||||||||||||
|
Total
|
Level 1
|
Level 2
|
Level 3
|
|||||||||||||
|
Assets:
|
||||||||||||||||
|
Certificates of Deposit
|
$
|
720,197
|
$
|
-
|
$
|
720,197
|
$
|
-
|
||||||||
|
Commercial Paper
|
3,985,740
|
-
|
3,985,740
|
-
|
||||||||||||
|
Corporate Bonds
|
4,031,170
|
-
|
4,031,170
|
-
|
||||||||||||
|
Total Assets:
|
$
|
8,737,107
|
$
|
-
|
$
|
8,737,107
|
$
|
-
|
||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrant Liabilities
|
$
|
1,573,366
|
$
|
-
|
$
|
-
|
$
|
1,573,366
|
||||||||
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
The fair value of the Company’s Level 2 marketable securities is determined by using quoted prices from independent pricing services that use market data for comparable securities in active or inactive markets. A variety of data inputs, including benchmark yields, interest rates, known historical trades and broker dealer quotes are using with pricing models to determine the quoted prices.
The fair value methodology for the warrant liabilities is disclosed in Note 12.
The carrying amounts reported in the financial statements for cash and cash equivalents (Level 1), and accounts payable and accrued expenses approximate fair value because of the short term maturity of these financial instruments.
The following table sets forth a reconciliation of changes in the years ended December 31, 2017 and 2016 in the fair value of the liabilities classified as Level 3 in the fair value hierarchy:
|
Warrant Liabilities
|
||||
|
Balance at January 1, 2017
|
$
|
1,573,366
|
||
|
Additions
|
6,738,701
|
|||
|
Unrealized losses, net
|
7,594,162
|
|||
|
Transfers out of level 3
|
(8,052,594
|
)
|
||
|
Balance at December 31, 2017
|
$
|
7,853,635
|
||
|
Warrant Liabilities
|
||||
|
Balance at January 1, 2016
|
$
|
2,739,163
|
||
|
Additions
|
4,364,110
|
|||
|
Unrealized gains, net
|
(5,529,907
|
)
|
||
|
Transfers out of level 3
|
-
|
|||
|
Balance at December 31, 2016
|
$
|
1,573,366
|
||
Additions consist of the fair value of warrant liabilities upon issuance. Transfers out of Level 3 for warrant liabilities consist of warrant exercises, where the liability is converted to additional paid-in capital upon exercise. The Company’s policy is to recognize transfers in and transfers out as of the actual date of the event or change in circumstance that caused the transfer.
REXAHN PHARMACEUTICALS, INC.
Notes to Financial Statements
| 17. |
Select Quarterly Data (Unaudited)
|
|
2017
|
||||||||||||||||
|
For the Quarter Ended
|
||||||||||||||||
|
March 31
|
June 30
|
September 30
|
December 31
|
|||||||||||||
|
Revenues
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Expenses
|
3,953,241
|
4,283,925
|
4,219,322
|
4,898,229
|
||||||||||||
|
Loss from Operations
|
(3,953,241
|
)
|
(4,283,925
|
)
|
(4,219,322
|
)
|
(4,898,229
|
)
|
||||||||
|
Other Income (Expense), net
|
(17,657,783
|
)
|
5,230,981
|
3,181,250
|
1,305,766
|
|||||||||||
|
Net Income (Loss)
|
$
|
(21,611,024
|
)
|
$
|
947,056
|
$
|
(1,038,072
|
)
|
$
|
(3,592,463
|
)
|
|||||
|
Net Income (Loss) per share, basic
|
$
|
(0.91
|
)
|
$
|
0.04
|
$
|
(0.04
|
)
|
$
|
(0.12
|
)
|
|||||
|
Net Income (Loss) per share, diluted
|
$
|
(0.91
|
)
|
$
|
0.03
|
$
|
(0.04
|
)
|
$
|
(0.12
|
)
|
|||||
|
2016
|
||||||||||||||||
|
For the Quarter Ended
|
||||||||||||||||
|
March 31
|
June 30
|
September 30
|
December 31
|
|||||||||||||
|
Revenues
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Expenses
|
4,863,981
|
3,912,782
|
3,717,575
|
3,919,047
|
||||||||||||
|
Loss from Operations
|
(4,863,981
|
)
|
(3,912,782
|
)
|
(3,717,575
|
)
|
(3,919,047
|
)
|
||||||||
|
Other Income, net
|
714,939
|
2,146,958
|
850,579
|
3,393,564
|
||||||||||||
|
Net Loss
|
$
|
(4,149,042
|
)
|
$
|
(1,765,824
|
)
|
$
|
(2,866,996
|
)
|
$
|
(525,483
|
)
|
||||
|
Net Loss per share, basic and diluted
|
$
|
(0.20
|
)
|
$
|
(0.08
|
)
|
$
|
(0.13
|
)
|
$
|
(0.02
|
)
|
||||
| 18. |
Subsequent Events
|
Since December 31, 2017, the Company granted 701,339 stock options to officers and other employees.
On February 5, 2018, the Company and NEXT BT Co. Ltd., the successor in interest to Rexgene, terminated the research collaboration agreement between the Company and Rexgene. In exchange for Next-BT terminating its rights to RX-0201 in Asia, the Company agreed to pay Next-BT a royalty in the low single digits of any net sales of RX-0201 the Company makes in Asia and 50% of the Company’s licensing revenue related to licensing of RX-0201 in Asia, up to an aggregate of $5,000,000.
On February 8, 2018, the Company entered into a research and development collaboration agreement with Zhejiang Haichang Biotechnology Co., Ltd. (“Haichang”) under which Haichang will develop a nano-liposomal formulation of RX-0201 using its proprietary QTzomes™ technology and will conduct certain pre-clinical and clinical activities through completion of a Phase 2a proof-of-concept clinical study in hepatocellular carcinoma in China.
F-33