10-K: Annual report pursuant to Section 13 and 15(d)
Published on
| ☒ |
Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
|
| ☐ |
Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
|
|
Delaware
|
11-3516358
|
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
|
37000 Grand River Avenue, Suite 1200
Farmington Hills, MI
|
48335
|
|
|
(Address of principal executive offices)
|
(Zip Code)
|
|
Title of each class
|
Trading Symbol
|
Name of each exchange on which registered
|
||
|
Common Stock, $0.0001 par value per share
|
OCUP
|
The Nasdaq Stock Market LLC
|
|
Large accelerated filer ☐
|
Accelerated filer ☐
|
||
|
Non-accelerated filer ☒
|
Smaller reporting company ☒
|
||
|
Emerging growth company ☐
|
|||
|
PART I
|
7
|
||
|
ITEM 1.
|
7
|
||
|
ITEM 1A.
|
68
|
||
|
ITEM 1B.
|
108
|
||
|
ITEM 2.
|
108
|
||
|
ITEM 3.
|
108
|
||
|
ITEM 4.
|
108
|
||
|
PART II
|
109
|
||
|
ITEM 5.
|
109
|
||
|
ITEM 6.
|
109
|
||
|
ITEM 7.
|
110
|
||
|
ITEM 7A.
|
124
|
||
|
ITEM 8.
|
124
|
||
|
ITEM 9A.
|
124
|
||
|
ITEM 9B.
|
125
|
||
|
PART III
|
126
|
||
|
ITEM 10.
|
126 |
||
|
ITEM 11.
|
EXECUTIVE COMPENSATION |
126
|
|
|
ITEM 12.
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
126
|
|
|
ITEM 13.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
126
|
|
|
ITEM 14.
|
PRINCIPAL ACCOUNTANT FEES AND SERVICES | 126 | |
|
|
|||
| PART IV | 127 | ||
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
127
|
|
|
ITEM 16.
|
131 | ||
|
159
|
|||
|
|
• |
Ocuphire currently depends entirely on the success of Nyxol and APX3330, its only product candidates. Ocuphire may never receive marketing approval for, or successfully commercialize, Nyxol, APX3330, or other
product candidates it may pursue in the future for any indication.
|
|
|
• |
The results of previous clinical trials may not be predictive of future results, and the results of Ocuphire’s current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory
authorities.
|
|
|
• |
Changes in regulatory requirements or FDA guidance, or unanticipated events during Ocuphire’s clinical trials, may result in changes to clinical trial protocols or additional clinical trial requirements, which could
result in increased costs to Ocuphire or delays in its development timeline.
|
|
|
• |
Ocuphire has incurred only losses since inception. Ocuphire expects to incur losses for the foreseeable future and may never achieve or maintain profitability.
|
|
|
• |
Ocuphire’s recurring operating losses have raised substantial doubt regarding its ability to continue as a going concern.
|
|
|
• |
Raising additional capital may cause dilution to Ocuphire’s stockholders, restrict Ocuphire’s operations, or require Ocuphire to relinquish rights to its technologies or product candidates.
|
|
|
• |
Even if it receives marketing approval for its product candidates in the United States, Ocuphire may never receive regulatory approval to market such product candidates outside of the United States.
|
|
|
• |
Even if Ocuphire obtains marketing approval for its product candidates, such product candidates could be subject to post-marketing restrictions or withdrawal from the market, and Ocuphire may be subject to
substantial penalties if it fails to comply with regulatory requirements or experience unanticipated problems with a product following approval.
|
|
|
• |
Ocuphire’s relationships with healthcare providers and third-party payors will be subject to applicable fraud and abuse and other healthcare laws and regulations, which could expose Ocuphire to criminal sanctions,
civil penalties, contractual damages, reputational harm, and diminished profits and future earnings, among other penalties and consequences.
|
|
|
• |
Ocuphire employees may engage in misconduct or other improper activities, including violating applicable regulatory standards and requirements or engaging in insider trading, which could significantly harm
Ocuphire’s business.
|
|
|
• |
Ocuphire faces substantial competition, which may result in others discovering, developing, or commercializing products before or more successfully than it does.
|
|
|
• |
Ocuphire lacks experience in commercializing products, which may have an adverse effect on its business.
|
|
|
• |
If Ocuphire is unable to establish sales and marketing capabilities or enter into agreements with third parties to sell, market, and distribute its product candidates, if approved, it may not be successful in
commercializing such product candidates if and when they are approved.
|
|
|
• |
Even if Ocuphire is able to commercialize its product candidates, their profitability will likely depend in significant part on third-party reimbursement practices, which, if unfavorable, would harm its business.
|
|
|
• |
Product liability lawsuits against Ocuphire, or its suppliers and manufacturers, could cause it to incur substantial liabilities and could limit commercialization of any product candidate that it may develop.
|
|
|
• |
Ocuphire will be unable to directly control all aspects of its clinical trials due to its reliance on clinical research organizations (“CROs”) and other third parties that assist Ocuphire in conducting clinical
trials.
|
|
|
• |
If Ocuphire is not able to establish new collaborations on commercially reasonable terms, it may have to alter its development, manufacturing, and commercialization plans.
|
|
|
• |
If Ocuphire is unable to obtain and maintain sufficient patent protection for its product candidates, its competitors could develop and commercialize products or technology similar or identical to those of Ocuphire,
which would adversely affect Ocuphire’s ability to successfully commercialize any product candidates it may develop, its business, results of operations, financial condition and prospects.
|
|
|
• |
If Ocuphire does not obtain protection under the Hatch-Waxman Act and similar foreign legislation by extending the patent terms and obtaining data exclusivity for its product candidate, its business may be
materially harmed.
|
|
|
• |
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing Ocuphire’s ability to protect its product candidates.
|
|
|
• |
Ocuphire may not be able to protect or practice its intellectual property rights throughout the world.
|
|
|
• |
Obtaining and maintaining Ocuphire’s patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by governmental agencies, and its patent
protection could be reduced or eliminated for noncompliance with these requirements.
|
|
|
• |
Ocuphire depends on intellectual property sublicensed from Apexian Pharmaceuticals, Inc. (“Apexian”) for its APX3330 product candidate under development and its additional pipeline candidates, and the termination
of, or reduction or loss of rights under, this sublicense would harm Ocuphire’s business.
|
|
|
• |
Ocuphire is dependent on its key personnel, and if it is not successful in attracting and retaining highly qualified personnel, it may not be able to successfully implement its business strategy.
|
|
|
• |
Ocuphire will need to develop and expand its company and may encounter difficulties in managing this development and expansion, which could disrupt its operations.
|
|
|
• |
The COVID-19 pandemic has and could continue to adversely impact Ocuphire’s business, including preclinical and clinical trials and regulatory approvals.
|
|
|
• |
Ocuphire’s insurance policies are expensive and protect only from some business risk, which leaves Ocuphire exposed to significant uninsured liabilities.
|
|
|
• |
If Ocuphire fails to comply with the continued listing standards of the Nasdaq Capital Market, Ocuphire common stock could be delisted. If it is delisted, Ocuphire common stock and the liquidity of its common stock
would be impacted.
|
|
|
• |
The market price of Ocuphire common stock may fluctuate significantly.
|
|
|
• |
Ocuphire may be subject to securities litigation, which is expensive and could divert management attention.
|
| ITEM 1. |
BUSINESS
|
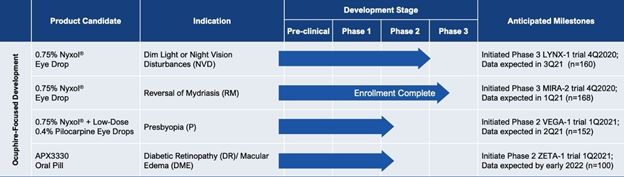
|
|
• |
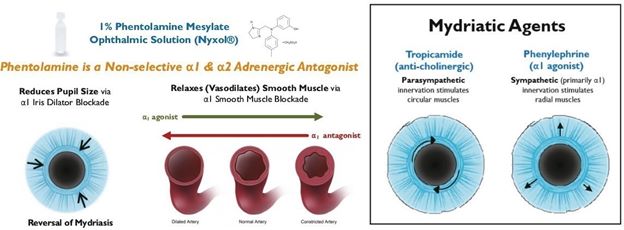
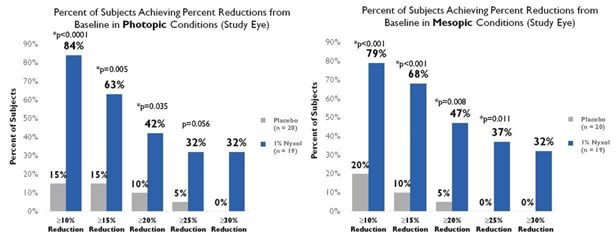
Reduction in pupil diameter with durable effects. In multiple Phase 2 trials Nyxol reduced pupil diameter by approximately 20% (~1 – 1.5 mm) in both mesopic (dim) and photopic
(bright) conditions, with such reductions sustained over 24 hours.
|
|
|
• |
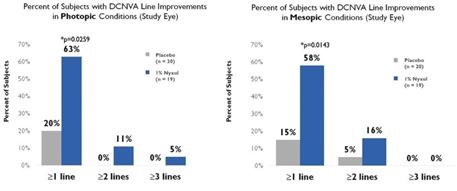
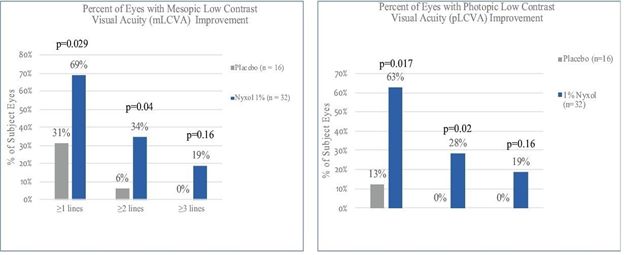
Improvement in low contrast visual acuity. When studied in patients with NVD in multiple Phase 2 trials, Nyxol showed statistically significant improvement in low contrast
mesopic best-corrected distance visual acuity at ≥1 and ≥2 lines, with a trend at ≥3 lines on a standard visual chart.
|
|
|
• |
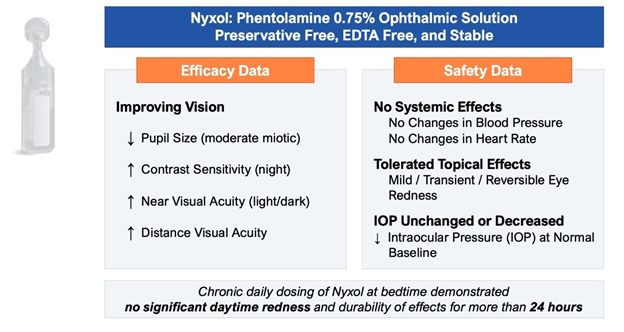
Promising tolerability profile. To date, Nyxol has been observed to be well tolerated, with unchanged or decreased intraocular pressure in the 7 completed Phase 1 and Phase 2
clinical trials conducted. Nyxol produces a transient, mild hyperemia effect that disappears within 4 to 8 hours or immediately upon application of anti-redness eye drops. Nyxol is also observed to have no systemic effects such as changes in
blood pressure or heart rate.
|
|
|
• |
Designed to be a convenient, once-daily eye drop. Nyxol is being evaluated for chronic use as a once-daily administration before bedtime. Nyxol has also been shown in multiple
Phase 2 trials to have an over 24-hour durable effect, which could allow for better patient compliance.
|
|
|
• |
Stable, cost-effective ophthalmic formulation. Nyxol is a single-use, preservative-free, proprietary eye drop formulation with good stability for eventual commercialization.
Its active pharmaceutical ingredient, phentolamine mesylate USP grade, is a small molecule with advantages of standardized, scalable, lower-cost manufacturing processes.
|
|
|
• |
NVD, a condition in which peripheral imperfections (aberrations) of the cornea scatter light when the pupil opens wide in dim light. Patients with NVD experience glare, halos,
starbursts, and decreased contrast sensitivity. NVD is a new indication with no approved therapies.
|
|
|
• |
RM, a reversal of pharmacologically induced dilation of the pupils, where dilation leads to increased sensitivity to light and an inability to focus, making it difficult to
read, work, and drive. RM is a single-use indication with no commercially available therapies.
|
|
|
• |
Presbyopia, a condition in which the eye’s lens loses elasticity, affecting its ability to focus on near objects. Presbyopia typically occurs after age 40 and most patients
use reading glasses in order to read or see objects close to them. There are no currently approved pharmacological therapies for presbyopia, but those in development plan to create a small pupil to better focus images on the retina via the
“pinhole effect”.
|
|
|
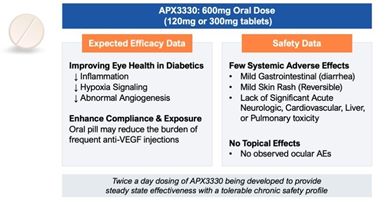
• |
Potential to be the first oral therapy. Compared to frequent intravitreal anti-VEGF injections, associated with ocular complications, twice a day oral administration of
APX3330 could be a convenient alternative treatment for retinal disease, if approved.
|
|
|
• |
Upstream target implicated in two validated pathways. APX3330 is designed to lead to inhibition of two validated cell signaling pathways (angiogenesis and inflammation) known
to cause various retinal diseases. Moreover, the APX3330 mechanism of action is distinct by working upstream of the current anti-VEGF therapies, thus Ocuphire believes it could complement anti-VEGF therapies and potentially reduce frequency
of doctor visits.
|
|
|
• |
Promising tolerability profile. In 11 completed Phase 1 and Phase 2 clinical trials, APX3330 was well tolerated with no significant acute neurologic, cardiovascular, liver, or
pulmonary events.
|
|
|
• |
Stable, cost-effective oral tablet. APX3330 is formulated as an oral tablet with stability suitable for eventual commercialization, and its active pharmaceutical ingredient
is a small molecule with the advantages of standardized, scalable, lower-cost manufacturing processes.
|
|
|
• |
DR, the leading cause of vision loss in adults aged 20–74 years, which results from chronic elevations of glucose in the blood that lead to cell damage in the retina.
|
|
|
• |
DME, one of the most common complications of DR, in which vascular leakage causes damage to the macula, the part of the eye that is critical for central and color vision.
|
|
|
• |
wAMD, a chronic eye disorder that causes visual distortions in the central part of one’s vision, in which abnormal blood vessels leak fluid or blood into the macula, the part
of the eye that is critical for central and color vision.
|
|
|
• |
Advance the clinical development of Nyxol and APX3330. Ocuphire is preparing to conduct registration studies of Nyxol and proof of concept studies of APX3330 with the
objective of filing a U.S. NDA in early 2023 for Nyxol and advancing APX3330 towards an NDA in the future.
|
|
|
• |
Target Nyxol and APX3330 for large ophthalmic indications. Ocuphire believes Nyxol has therapeutic potential to improve vision performance in NVD, RM, and presbyopia. Ocuphire
also believes AXP3330 has potential to improve the health of the retina in patients with diabetic retinopathy, diabetic macular edema, and wAMD, while reducing the burden of intravitreal injections.
|
|
|
• |
Maintain and expand its intellectual property portfolio. Ocuphire owns all global patent rights to Nyxol with respect to its formulation, combinations, and use in multiple
indications. Ocuphire also owns an exclusive worldwide sublicense for the Ref-1 Inhibitor program, including its lead product candidate APX3330, for all its ophthalmic and diabetic indications, and compositions and methods of use for Ref-1
pipeline candidates, including APX2009 and APX2014. Ocuphire continues to explore additional opportunities to expand and extend this intellectual property protection, both in the U.S. and in other jurisdictions.
|
|
|
• |
Maximize the global commercial value of Nyxol and APX3330. Ocuphire plans to seek commercial partners both in and outside of the United States. Alternatively, Ocuphire
believes it could independently commercialize Nyxol and/or APX3330 in the United States with a targeted sales force.
|
|
|
• |
Evaluate in-licensing and acquisition opportunities. Ocuphire’s team is well qualified to identify and in-license or acquire clinical-stage ophthalmological assets and is
evaluating opportunities to expand and diversify its pipeline.
|
Vision at night or in dim light conditions is different from daytime vision in several important ways. Most notably, at night, the pupils dilate to allow more light into the eye. Diminished night vison is a natural part of aging as well as a common side effect of several conditions and procedures. NVD is caused by peripheral imperfections (aberrations) of the cornea which scatter light when the pupil dilates in dim light conditions. These imperfections can be naturally occurring, especially with age, or surgically-induced from refractive procedures such as LASIK. As the pupil dilates in response to mesopic conditions, light passes through the periphery of the cornea and lens, unlike during photopic conditions. Any imperfections or aberrations present on the periphery cause light to reach the retina in a non-focused and scattered way, creating glare, halos, starbursts, ghosting, and a loss of contrast sensitivity (“CS”). These visual disturbances can be debilitating to a variety of everyday activities, especially driving. The light emitted by traffic lights and other cars scatters and obscures most of the visual field, making driving in dim light conditions hazardous. Glare, in particular, can be dangerous while driving. In one study of 297 drivers given vision tests that correlate with accidents, 45% of the drivers who reported difficulty driving at night were unable to perform any of the tests with glare.
|
|
• |
Non-proliferative DR, or NPDR. NPDR is an earlier, more typical stage of DR and can progress into more severe forms of DR over time if untreated and if exposure to elevated
blood sugar levels persists.
|
|
|
• |
Proliferative DR, or PDR. PDR is a more advanced stage of DR than NPDR. It is characterized by retinal neovascularization and, if left untreated, leads to permanent damage
and blindness.
|
|
|
• |
In a double-masked, randomized, single dose, 3-arm controlled, parallel design Phase 1 trial (OP-NYX-001, IND 67-288), 45 healthy volunteers were administered a single dose of 0.2% Nyxol with or without
tetrahydrozoline or tetrahydrozoline alone. Both Nyxol-treated groups showed a statistically significant reduction in pupil diameter (PD) compared to tetrahydrozoline alone.
|
|
|
• |
In a 12-day, double-masked, randomized, placebo-controlled, single-dose, incomplete block, 3-period crossover, dose escalation Phase 1 trial in 16 healthy volunteers (OP-NYX-002, IND 67-288), there was a
dose-related response in improvement in LCVA relative to placebo.
|
|
|
• |
In a 2-week, double-masked, randomized, placebo-controlled, single-dose, incomplete block 3-period crossover, dose escalation Phase 1/2 trial in 16 patients with NVD (OP-NYX-004, IND 73-987), Nyxol was
well-tolerated with no severe adverse events (SAEs).
|
|
|
• |
In a 1-day, double-masked, randomized, placebo-controlled, single-dose Phase 2 trial in 24 patients with severe NVD (OP-NYX-SNV, IND 70-736), patients treated with Nyxol exhibited greater reductions in pupil
diameter and greater improvements in low contrast visual acuity compared to those on placebo.
|
|
|
• |
In a 15-day, double-masked, randomized, placebo-controlled, multiple-dose, 3-arm (0, 0.5%, and 1% Nyxol) Phase 2 trial in 60 patients with severe NVD (OP-NYX-01a2, IND 70499), improvements in contrast sensitivity
frequencies and VA, as well as reductions in intraocular pressure (IOP) and pupil diameter, were observed.
|
|
|
• |
In a 14-day, double-masked, randomized, placebo-controlled, multiple-dose, multi-center Phase 2b trial in 39 patients with elevated intraocular pressure (ORION-1, IND 070499), patients treated with 1% Nyxol showed
statistically significant reduction in PD and improvement in near visual acuity relative to placebo, with evening bedtime daily dosing regimen.
|
|
|
• |
In a double-masked, randomized, placebo-controlled, crossover, single-dose, multi-center Phase 2b trial with 32 healthy patients (MIRA-1, IND 070499) to study reversal of pharmacologically induced mydriasis, healthy
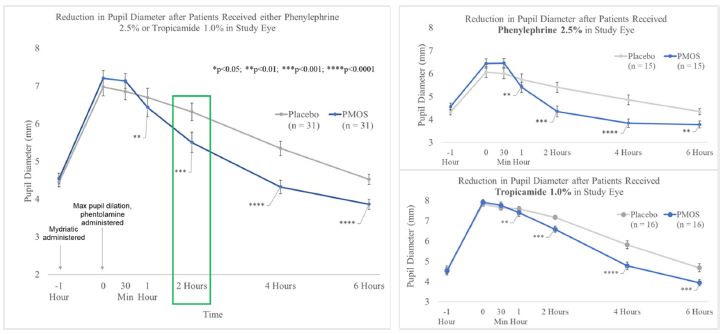
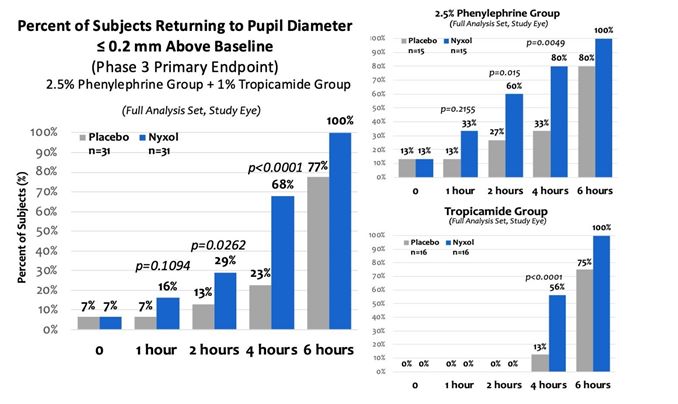
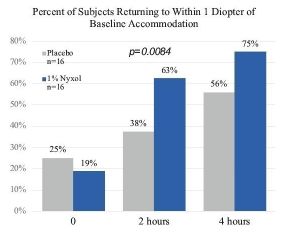
patients treated with 1% Nyxol had statistically significantly greater reductions in PD at multiple time points compared to placebo, and more patients in the study group returned to baseline PD at 2 hours compared to the placebo group.
|
|
Trial
Name (IND
Number)
|
|
Patient /
Indication
|
|
Phase
|
|
Trial Objectives
|
|
Doses
|
|
Number of
Patients^
|
|
Dosing
|
|
Key
Endpoints
|
|
NYX-001
(67-288)
|
|
Healthy Volunteers
|
|
1
|
|
Double-masked, randomized, single dose, 3-arm controlled, parallel trial to determine the efficacy and safety of phentolamine mesylate
|
|
0.2%
|
|
Nyxol*=15, Visine=15,
Visine + Nyxol*=15
Total = 45
|
|
Single-dose
|
|
Safety and Efficacy (PD)
|
|
NYX-002
(67-288)
|
|
Healthy Volunteers
|
|
1
|
|
Double-masked, randomized, placebo-controlled, single-dose, incomplete block, 3-period crossover, dose escalation trial evaluating the tolerability and efficacy of phentolamine mesylate
|
|
0.2%, 0.4%, 0.8%
|
|
Nyxol*=16 Placebo=12
Total = 16
|
|
Single-dose
|
|
Safety and Efficacy (PD, VA)
|
|
OP-NYX-004
(73-987)
|
|
Night Vision Disturbances Patients
|
|
1 / 2
|
|
Double-masked, randomized, placebo-controlled, single-dose, incomplete block 3-period crossover, dose escalation trial to determine the efficacy and safety of phentolamine mesylate
|
|
0.2%, 0.4%, 0.8%
|
|
Nyxol*=16 Placebo=12
Total = 16
|
|
Single-dose
|
|
Safety and Efficacy
|
|
OP-NYX-SNV
(70-736)
|
|
Severe Night Vision Disturbances Patients
|
|
2
|
|
Double-masked, randomized, placebo-controlled, single-dose trial to assess the efficacy and safety of phentolamine mesylate ophthalmic solution
|
|
1.0%
|
|
Nyxol*=16, Placebo=8
Total = 24
|
|
Single-dose
|
|
Safety and Efficacy (PD, LCVA, CS, WA)
|
|
OP-NYX-01a2
(70-499)
|
|
Severe Night Vision Disturbances Patients
|
|
2
|
|
Double-masked, randomized, placebo-controlled, single-dose, 3-arm trial to assess the efficacy and safety of Nyxol
|
|
0.5%, 1.0%
|
|
Nyxol=40 Placebo=20
Total = 60
|
|
Multiple doses (15-28 days)
|
|
Safety and Efficacy (PD, LCVA, CS)
|
|
OPI-NYXG-201
(ORION-1)
(70-499)
|
|
Glaucoma and Ocular Hypertension, Elderly Patients
|
|
2b
|
|
Double-masked, randomized, placebo-controlled, multiple-dose, multi-center trial to assess the efficacy and safety of Nyxol
|
|
1.0%
|
|
Nyxol=19 Placebo=20
Total = 39
|
|
Multiple doses (14 days)
|
|
Safety and Efficacy (IOP, PD, near VA, VA)
|
|
OPI-
NYXRM-201
(MIRA-1)
(70-499)
|
|
Healthy Patients/ Reversal of Mydriasis
|
|
2b
|
|
Double-masked, randomized, placebo-controlled, crossover, single-dose, multi-center trial to assess the efficacy and safety of Nyxol in reducing pharmacologically induced
mydriasis
|
|
1.0%
|
|
Nyxol=31 Placebo=32
Total = 32
|
|
Single-dose
|
|
Safety and Efficacy (PD, Accommodation, VA)
|
|
Study
|
|
Group
|
|
Mesopic Conditions
|
||||||||
|
|
Pre-Treatment
(Baseline) Pupil
Diameter
|
|
Post-
Treatment
Pupil
Diameter
|
|
Change (%)
|
|
p-value
compared
to baseline
|
|
p-value
compared
to placebo
|
|||
|
NYX-SNV
|
|
Placebo (N = 16)
|
|
6.6mm
|
|
6.4mm
|
|
-0.2mm (-3%)
|
|
p = 0.08
|
|
p < 0.0001
|
|
|
1% Nyxol (N = 32)
|
|
6.5mm
|
|
5.2mm
|
|
-1.3mm (-20%)
|
|
p < 0.0001
|
|
||
|
NYX-01a2
|
|
Placebo (N = 38)
|
|
6.25mm
|
|
6.31mm
|
|
0.07mm (+1%)
|
|
p = 0.6
|
|
p < 0.0001
|
|
|
1% Nyxol (N = 40)
|
|
6.17mm
|
|
5.31mm
|
|
-0.86mm (-14%)
|
|
p < 0.0001
|
|
||
|
NYXG-201
|
|
Placebo (N = 20)
|
|
4.57mm
|
|
4.52mm
|
|
-0.05mm (-1%)
|
|
p = 0.6178
|
|
p < 0.0001
|
|
|
1% Nyxol (N = 19)
|
|
4.69mm
|
|
3.70mm
|
|
-1.00mm (-21%)
|
|
p < 0.0001
|
|
||
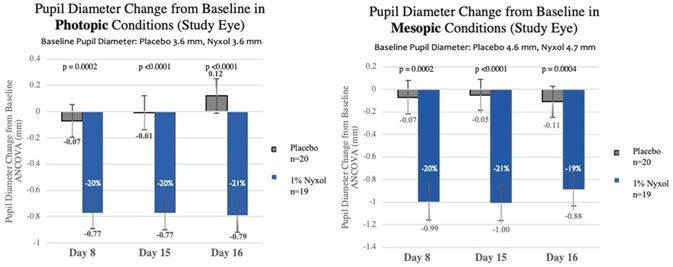
|
System Organ Class
Preferred Term
|
|
Nyxol
(n=19)
n (%)
|
|
Placebo
(n=20)
n (%)
|
|
Total number of TEAEs, n[1]
|
|
16
|
|
2
|
|
Eye disorders
|
|
3 (15.8)
|
|
1 (5.0)
|
|
Conjunctival hyperemia
|
|
3 (15.8)
|
|
1 (5.0)
|
|
Eye pruritus
|
|
1 (5.3)
|
|
0
|
|
Vision blurred
|
|
0
|
|
0
|
|
Conjunctival hemorrhage
|
|
0
|
|
0
|
|
Corneal deposits
|
|
0
|
|
0
|
|
Erythema of eyelid
|
|
0
|
|
0
|
|
Eye irritation
|
|
0
|
|
0
|
|
Eyelid edema
|
|
0
|
|
0
|
|
System Organ Class
Preferred Term
|
|
Nyxol
(n=19)
n (%)
|
|
Placebo
(n=20)
n (%)
|
|
Lacrimation increased
|
|
0
|
|
0
|
|
Eye pain
|
|
0
|
|
0
|
|
Visual acuity reduced
|
|
0
|
|
0
|
|
Conjunctival edema
|
|
0
|
|
0
|
|
Foreign body sensation in eyes
|
|
0
|
|
0
|
|
Punctate keratitis
|
|
0
|
|
0
|
|
General disorders and administration site conditions
|
|
3 (15.8)
|
|
0
|
|
Instillation site burn
|
|
2 (10.5)
|
|
0
|
|
Instillation site pain
|
|
1 (5.3)
|
|
0
|
|
Infections and infestations
|
|
1 (5.3)
|
|
0
|
|
Prostate infection
|
|
1 (5.3)
|
|
0
|
|
Upper respiratory tract infection
|
|
1 (5.3)
|
|
0
|
|
Nervous system disorders
|
|
0
|
|
0
|
|
Headache
|
|
0
|
|
0
|
|
Skin and subcutaneous tissue disorders
|
|
0
|
|
0
|
|
Injury, poisoning and procedural complication
|
|
0
|
|
0
|
|
Respiratory, thoracic, and mediastinal disorders
|
|
0
|
|
0
|
|
Cardiac disorders
|
|
0
|
|
0
|
|
Vascular disorders
|
|
0
|
|
0
|
|
Variable
|
|
Placebo
(N = 40)
|
|
0.5% Nyxol
(N = 40)
|
|
1% Nyxol
(N = 40)
|
|
Pre-Treatment Day 1 IOP (mmHg ± STDEV)
|
|
16.1 ± 2.3
|
|
16.7 ± 2.7
|
|
16.6 ± 2.5
|
|
Post-Treatment Day 1 IOP (mmHg ± STDEV)
|
|
16.2 ± 3.2
|
|
15.4 ± 3.6
|
|
14.2 ± 2.9
|
|
Change from Pretreatment Day 1 IOP (mmHg ± STDEV)
|
|
0.1 ± 2.7
|
|
-1.3 ± 3.2
|
|
-2.4 ± 2.2
|
|
Change in Baseline Significance^
|
|
p = 0.9192
|
|
p = 0.0043
|
|
p < 0.0001
|
|
Change compared to Placebo Significance^
|
|
N/A
|
|
p = 0.0148
|
|
p < 0.0001
|
| ^ |
P-values were generated using the Wilcoxon Signed Rank Test.
|
|
|
• |
APX_CLN_0001: A Phase 1, randomized, single-dose placebo-controlled trial of APX3330 to investigate the safety and pharmacokinetics during oral dosing of APX3330 to healthy
adult males. A total of 18 patients were treated with single oral doses of APX3330 (10 mg, 30 mg, 60 mg, 120 mg, 180 mg or 240 mg) or the placebo in a blind manner.
|
|
|
• |
APX_CLN_0002: An 8-day, randomized Phase 1 repeat-dose placebo-controlled trial to investigate the safety and pharmacokinetics of orally dosed APX3330 in healthy adult male
patients. A total of 18 patients were treated with oral dosing of APX3330 (120 mg or 240 mg) or the placebo in a blind manner once or twice a day for 8 successive days.
|
|
|
• |
APX_CLN_0003: A 7-day Phase 1 repeat-dose trial (120 mg) in 6 healthy patients to determine the effects of food on orally administered APX3330.
|
|
|
• |
APX_CLN_0004 A single-dose trial (120 mg) in 6 healthy patients to determine the effect of meals on the pharmacokinetics of APX3330.
|
|
|
• |
APX_CLN_0005 A 12-week dose-escalation Phase 2 trial (20 mg, 60 mg, 120 mg, 240 mg) in 40 chronic hepatitis B patients. Patients received oral administration of one tablet per
dose (2 tablets in the case of the administration of 240 mg) twice a day, after breakfast and after dinner.
|
|
|
• |
APX_CLN_0006 A 12-week dose-escalation Phase 2 trial (20 mg, 60 mg, 120 mg, 240 mg) in 51 chronic hepatitis C patients. The objective of the trial was to investigate the
safety, efficacy and utility of APX3330 in treating patients with chronic hepatitis C.
|
|
|
• |
APX_CLN_0007 A 12-week double-masked, randomized placebo-controlled Phase 2 trial (0 mg, 120 mg, 240 mg) in chronic hepatitis C patients that had failed previous interferon
treatment. Safety was evaluated in 196 completed patients. The mean treatment period in each group was 82 days in the placebo group, 79 days in the 120 mg group and 78 days in the 240 mg group. The primary endpoints of this trial were
measurement of the rate of change in the glutamic pyruvate transaminase (GPT) level, degree of improvement in liver function and assessment of general performance status.
|
|
|
• |
APX_CLN_0008 A 3-step, Phase 1 single-dose, single-blind trial (300 mg, 420 mg, 600 mg) in 27 healthy patients to investigate the safety and pharmacokinetics of higher doses.
|
|
|
• |
APX_CLN_0009 A 2-week repeated-dose Phase 2 trial (120 mg) in 30 patients with acute severe hepatitis, including patients with advanced liver cirrhosis. Efficacy endpoints
included objective measures of liver function and subjective improvement of patient functional status. Safety measures included the assessment of the general tolerability of the drug (i.e., changes in vital signs) and changes in clinical
laboratory values.
|
|
|
• |
APX_CLN_00010 A 4-week repeated-dose Phase 2 trial (120 mg) in 30 patients with alcoholic hepatitis, including patients with liver cirrhosis. Efficacy endpoints included
objective measures of liver function and subjective improvement of patient functional status. Safety measures included the assessment of the general tolerability of the product candidate (i.e., changes in vital signs) and changes in clinical
laboratory values.
|
|
|
• |
APX_CLN_0011 was a multi-center, open-label, dose-escalation Phase 1 oncology trial in patients with advanced solid tumors. Patients received daily oral doses of APX3330 each
day of repeated 21-day cycles until disease progression or trial withdrawal. Nineteen patients received APX3330 in escalating doses from 240 mg/d dose to 720 mg/d in increments of 120mg/d. The top dose tested (720 mg/d) produced a
self-limiting, diffuse macular rash and was confirmed as the dose-limiting toxicity. The dose of 600 mg/d was then confirmed as a dose tolerable for chronic administration and for further clinical development as a modulator of Ref-1 activity
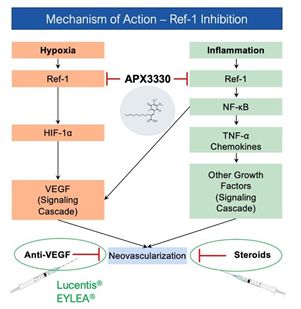
in inflammatory diseases. Biopsy analyses of patients participating in the trial confirmed that APX3330 directly targets the Ref-1 protein and that the targeting produces subsequent regulation of transcription factors such as NF-κB and
HIF-1α, regulators of VEGF and other inflammatory molecules. This mechanism of action provides significant rationale for testing APX3330 in diseases in which inflammation and neo-vascular development play a critical pathogenic role.
|
|
Trial Number /
Name
|
|
Patient /
Indication
|
|
Phase
|
|
Trial Objectives
|
|
Doses
|
|
Number
of
Patients
|
|
Dosing
|
|
Key Endpoints
|
|
APX_CLN_0001
|
|
Healthy Volunteers
|
|
1
|
|
Single-dose placebo-controlled trial of APX3330 to investigate safety and pharmacokinetics
|
|
10 mg 30 mg 60 mg 120 mg 180 mg 240 mg
|
|
APX3330 = 9 Placebo = 9
|
|
Single dose
|
|
Plasma Concentration of total quinone forms, safety
|
|
APX_CLN_0002
|
|
Healthy Volunteers
|
|
1
|
|
Repeat-dose placebo-controlled trial to investigate safety and pharmacokinetics
|
|
120 mg QD 120 mg BID
|
|
APX3330 = 9 Placebo = 9
|
|
8 days
|
|
Plasma Concentration of APX3330, safety
|
|
APX_CLN_0003
|
|
Healthy Volunteers
|
|
1
|
|
Repeat-dose trial to determine effects of food on pharmacokinetics
|
|
240 mg
|
|
APX3330 = 6
|
|
1 week
|
|
Plasma Concentration of APX3330, safety
|
|
APX_CLN_0004
|
|
Healthy Volunteers
|
|
1
|
|
Single-dose trial to determine the effects of meals on pharmacokinetics
|
|
120 mg
|
|
APX3330 = 6
|
|
Single dose
|
|
Plasma Concentration of APX3330, Safety
|
|
APX_CLN_0005
|
|
Chronic Hepatitis B Patients
|
|
2
|
|
Dose-escalation trial to investigate safety, efficacy and tolerability
|
|
20 mg 60 mg 120 mg 240 mg
|
|
APX3330 = 40
|
|
12 weeks
|
|
Safety
|
|
APX_CLN_0006
|
|
Chronic Hepatitis C Patients
|
|
2
|
|
Dose-escalation trial to investigate safety, efficacy and tolerability
|
|
20 mg 60 mg 120 mg 240 mg
|
|
APX3330 = 51
|
|
12 weeks
|
|
Safety
|
|
Trial Number /
Name
|
|
Patient /
Indication
|
|
Phase
|
|
Trial Objectives
|
|
Doses
|
|
Number
of
Patients
|
|
Dosing
|
|
Key Endpoints
|
|
APX_CLN_0007
|
|
Chronic Hepatitis C Patients
|
|
2
|
|
Double-masked, placebo-controlled trial to investigate safety, efficacy and tolerability
|
|
120 mg 240 mg
|
|
APX3330 = 128
Placebo = 68
|
|
Placebo = 82 days
APX3330 120 mg = 79 days
240 mg = 78 days
|
|
Rate of change in GPT level, improvement in liver function, general performance
|
|
APX_CLN_0008
|
|
Healthy Patients
|
|
1
|
|
Single-blind, single-dose, 3-step trial to investigate safety and pharmacokinetics of higher doses
|
|
300 mg 420 mg 600 mg
|
|
APX3330 = 27
|
|
Single dose
|
|
Plasma Concentration of APX3330, safety
|
|
APX_CLN_0009
|
|
Advanced Liver Cirrhosis Patients
|
|
2
|
|
Repeated-dose trial to investigate safety, efficacy and tolerability
|
|
120 mg
|
|
APX3330 = 30
|
|
2 weeks
|
|
Liver function, patient functional status, tolerability
|
|
APX_CLN_0010
|
|
Advanced Liver Cirrhosis Patients
|
|
2
|
|
Repeated-dose trial to investigate safety, efficacy and tolerability
|
|
120 mg
|
|
APX3330 = 30
|
|
4 weeks
|
|
Liver function, patient functional status, tolerability
|
|
APX_CLN_0011
|
|
Advanced Solid Tumor Patients
|
|
1
|
|
Multicenter, open-label, dose-escalation to investigate safety, efficacy, pharmacokinetics, and recommended Phase 2 dose
|
|
240 mg 360 mg 480 mg 600 mg 720 mg
|
|
APX3330 = 19
|
|
21-day cycles until disease progression or study withdrawal
|
|
Tumor response, safety, PK, target engagement
|
|
System Organ Class
Preferred Term
|
|
|
APX3330
20-240 mg
(N=279)
|
|
|
Placebo
(N=68)
|
||||||
|
|
n (%)
|
|
|
# events
|
|
|
n (%)
|
|
|
# events
|
||
|
Adverse Events
|
|
|
40 (14.3)
|
|
|
52
|
|
|
11 (16.2)
|
|
|
15
|
|
Blood and lymphatic system disorders
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Anemia
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Cardiac disorders
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Palpitations
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Gastrointestinal disorders
|
|
|
12 (4.3)
|
|
|
14
|
|
|
2 (2.9)
|
|
|
2
|
|
Abdominal discomfort
|
|
|
1 (0.4)
|
|
|
1
|
|
|
1 (1.5)
|
|
|
1
|
|
Abdominal pain
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Abdominal pain lower
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Cheilitis
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Diarrhea
|
|
|
3 (1.1)
|
|
|
3
|
|
|
0
|
|
|
0
|
|
Feces soft
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Gastric ulcer
|
|
|
2 (0.7)
|
|
|
2
|
|
|
0
|
|
|
0
|
|
Hypo aesthesia oral
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Mouth swelling
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Stomatitis
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Tongue dry
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Vomiting
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
General disorders and administration site conditions
|
|
|
6 (2.2)
|
|
|
6
|
|
|
3 (4.4)
|
|
|
3
|
|
Chest discomfort
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Feeling abnormal
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Malaise
|
|
|
3 (1.1)
|
|
|
3
|
|
|
1 (1.5)
|
|
|
1
|
|
Peripheral edema
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Peripheral swelling
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Pyrexia
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Infections and infestations
|
|
|
3 (1.1)
|
|
|
3
|
|
|
0
|
|
|
0
|
|
Nasopharyngitis
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Upper respiratory tract infections
|
|
|
2 (0.7)
|
|
|
2
|
|
|
0
|
|
|
0
|
|
Investigations
|
|
|
2 (0.7)
|
|
|
2
|
|
|
0
|
|
|
0
|
|
Blood urea increased
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Urobilinogen urine increased
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Musculoskeletal and connective tissue disorders
|
|
|
0
|
|
|
0
|
|
|
2 (2.9)
|
|
|
3
|
|
Limb discomfort
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Musculoskeletal pain
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Pain in extremity
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Nervous system disorders
|
|
|
4 (1.4)
|
|
|
6
|
|
|
4 (5.9)
|
|
|
5
|
|
Ageusia
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Burning sensation
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Dizziness
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Headache
|
|
|
2 (0.7)
|
|
|
2
|
|
|
1 (1.5)
|
|
|
1
|
|
Hypoaesthesia
|
|
|
1 (0.4)
|
|
|
1
|
|
|
1 (1.5)
|
|
|
1
|
|
Hypoglycemic coma
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Parosmia
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Subarachnoid hemorrhage
|
|
|
0
|
|
|
0
|
|
|
1 (1.5)
|
|
|
1
|
|
Eye disorders
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Ocular discomfort
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Psychiatric disorders
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Insomnia
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Renal and urinary disorders
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Hematuria
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Respiratory, thoracic and mediastinal disorders
|
|
|
2 (0.7)
|
|
|
2
|
|
|
1 (1.5)
|
|
|
1
|
|
Acute respiratory distress syndrome
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Upper respiratory tract inflammation
|
|
|
1 (0.4)
|
|
|
1
|
|
|
1 (1.5)
|
|
|
1
|
|
System Organ Class
Preferred Term
|
|
|
APX3330
20-240 mg
(N=279)
|
|
|
Placebo
(N=68)
|
||||||
|
|
n (%)
|
|
|
#
events
|
|
|
n (%)
|
|
|
#
events
|
||
|
Skin and subcutaneous tissue disorders
|
|
|
12 (4.3)
|
|
|
14
|
|
|
1 (1.5)
|
|
|
1
|
|
Alopecia
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Drug eruption
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Dry skin
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Eczema
|
|
|
2 (0.7)
|
|
|
2
|
|
|
0
|
|
|
0
|
|
Papule
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
|
Pruritus
|
|
|
5 (1.8))
|
|
|
5
|
|
|
1 (1.5)
|
|
|
1
|
|
Rash
|
|
|
2 (0.7)
|
|
|
2
|
|
|
0
|
|
|
0
|
|
Urticaria
|
|
|
1 (0.4)
|
|
|
1
|
|
|
0
|
|
|
0
|
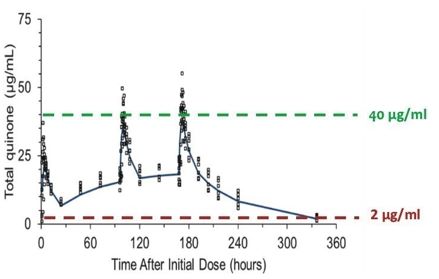
|
|
• |
Liquid Vision®, with aceclidine (another miotic agent), developed by Presbyopia Therapies, LLC.
|
|
|
• |
MicroLine®, which is a microdose formulation of pilocarpine, developed by Eyenovia, Inc.
|
|
|
• |
KT-101, which uses pilocarpine in the AcuStream delivery system, developed by Kedalion Therapeutics, Inc.
|
|
|
• |
BrimocholTM, with brimonidine and carbachol (both are miotic agents), developed by Visus Therapeutics, Inc.
|
|
|
• |
UNR844, which uses a mechanism that involves softening the lens to increase near visual acuity, developed by Novartis AG (originally Encore Vision, Inc.).
|
|
|
• |
Lucentis® (ranibizumab) and Avastin® (bevacizumab), which are anti-VEGF monoclonal antibody intravitreal injections, developed by Genentech, Inc.
|
|
|
• |
EYLEA® (aflibercept), a VEGF inhibitor intravitreal injection, developed by Regeneron Pharmaceuticals.
|
|
|
• |
Beovu® Brolucizumab, an anti-VEGF monoclonal antibody intravitreal injection, developed by Novartis AG.
|
|
|
• |
MACUGEN® (pegaptanib sodium injection), a selective inhibitor of VEGF-165, developed by Bausch + Lomb.
|
|
|
• |
Ozurdex® (dexamethasone), a corticosteroid IVT implant, developed by Allergan plc.
|
|
|
• |
Iluvien (fluocinolone acetonide), a corticosteroid IVT implant, developed by Alimera Sciences, Inc.
|
|
|
• |
Abicipar, an anti-VEGF intravitreal injection with a long duration of action, developed by Allergan plc and Molecular Partners.
|
|
|
• |
Farcimab, a bispecific antibody intravitreal injection that suppresses both VEGF and Angiopoietin-2, developed by Genentech, Inc. and Roche AG.
|
|
|
• |
KSI-301, an anti-VEGF antibody intravitreal injection coupled with a biopolymer that is intended to increase the time between injections, developed by Kodiak Sciences.
|
|
|
• |
OPT-302, an intravitreal injection which binds to multiple types of VEGF receptors that could be used with other anti-VEGF agents, developed by Opthea Limited.
|
|
|
• |
ALG-1001, an integrin peptide therapy intravitreal injection that is being evaluated as a sequential or in-combination therapy with bevacizumab in patients with DME, developed by Allegro Ophthalmics, LLC.
|
|
|
• |
completion of preclinical laboratory tests, animal studies and formulation studies in compliance, as applicable, with the Animal Welfare Act and FDA’s good laboratory practice, or GLP, regulations;
|
|
|
• |
submission to the FDA of an IND, which must take effect before human clinical trials may begin;
|
|
|
• |
approval by an independent institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated;
|
|
|
• |
performance of adequate and well-controlled human clinical trials in accordance with good clinical practices, or GCP, to establish the safety and efficacy of the proposed drug product for each indication;
|
|
|
• |
preparation and submission to the FDA of an NDA;
|
|
|
• |
review of the product by an FDA advisory committee, where appropriate or if applicable;
|
|
|
• |
satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with current Good Manufacturing Practices, or cGMP,
requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity;
|
|
|
• |
satisfactory completion of FDA audits of clinical trial sites to assure compliance with GCPs and the integrity of the clinical data;
|
|
|
• |
payment of user fees and securing FDA approval of the NDA; and
|
|
|
• |
compliance with any post-approval requirements, including Risk Evaluation and Mitigation Strategies, or REMS, and post-approval studies required by the FDA.
|
|
|
• |
Phase 1. The drug is initially introduced into healthy human patients or, in certain indications such as cancer, patients with the target disease or condition and tested for safety, dosage
tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage.
|
|
|
• |
Phase 2. The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted
diseases and to determine dosage tolerance and optimal dosage.
|
|
|
• |
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
|
|
|
• |
fines, warning letters or holds on post-approval clinical trials;
|
|
|
• |
refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals;
|
|
|
• |
product seizure or detention, or refusal to permit the import or export of products; or
|
|
|
• |
injunctions or the imposition of civil or criminal penalties.
|
|
|
• |
the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or
reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid;
|
|
|
• |
the federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalties laws, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented,
to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
|
|
|
• |
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal laws that prohibit, among other things, knowingly and willingly executing, or attempting to execute, a scheme to
defraud any healthcare benefit program or making false statements relating to healthcare matters;
|
|
|
• |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, which also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy,
security and transmission of individually identifiable health information;
|
|
|
• |
the federal transparency requirements known as the federal Physician Payments Sunshine Act, under the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, or the Affordable Care Act, which
requires certain manufacturers of drugs, devices, biologics and medical supplies to report specially to the Centers for Medicare & Medicaid Services, or CMS, within the U.S. Department of Health and Human Services, information related to
payments and other transfers of value to clinicians and teaching hospitals and clinician ownership and investment interests; and
|
|
|
• |
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers.
|
|
|
• |
a special, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs,
although this fee would not apply to sales of certain products approved exclusively for orphan indications;
|
|
|
• |
expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a
manufacturer’s Medicaid rebate liability;
|
|
|
• |
expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs and revising the definition of “average manufacturer price,” or AMP, for calculating and
reporting Medicaid drug rebates on outpatient prescription drug prices and extending rebate liability to prescriptions for individuals enrolled in Medicare Advantage plans; addressed a new methodology by which rebates owed by manufacturers
under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
|
|
|
• |
expanded the types of entities eligible for the 340B drug discount program;
|
|
|
• |
established the Medicare Part D coverage gap discount program by requiring manufacturers to provide a 50% point-of-sale-discount off the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period
as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D;
|
|
|
• |
a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research;
|
|
|
• |
the Independent Payment Advisory Board, or IPAB, which has authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription drugs. However, the IPAB
implementation has been not been clearly defined. The ACA provided that under certain circumstances, IPAB recommendations will become law unless Congress enacts legislation that will achieve the same or greater Medicare cost savings; and
|
|
|
• |
established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. Funding has been
allocated to support the mission of the Center for Medicare and Medicaid Innovation from 2011 to 2019.
|
| ITEM 1A. |
|
|
• |
the data collected from preclinical studies and clinical trials of Ocuphire’s product candidates may not be sufficient to support the submission of an NDA;
|
|
|
• |
Ocuphire may not be able to demonstrate to the satisfaction of the FDA that its product candidates are safe and effective for any indication;
|
|
|
• |
the results of clinical trials may not meet the level of statistical significance or clinical significance required by the FDA for approval;
|
|
|
• |
the FDA may disagree with the number, design, size, conduct, or implementation of Ocuphire’s clinical trials;
|
|
|
• |
the FDA may not find the data from preclinical studies and clinical trials sufficient to demonstrate that Ocuphire’s product candidates’ clinical and other benefits outweigh the safety risks;
|
|
|
• |
the FDA may disagree with Ocuphire’s interpretation of data from preclinical studies or clinical trials;
|
|
|
• |
the FDA may not accept data generated at Ocuphire’s clinical trial sites;
|
|
|
• |
the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of Ocuphire’s application or may recommend that the FDA require, as a condition of
approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions;
|
|
|
• |
the FDA may require development of a Risk Evaluation and Mitigation Strategy (REMS) as a condition of approval;
|
|
|
• |
the FDA may identify deficiencies in the manufacturing processes or facilities of third-party manufacturers with which Ocuphire enters into agreements for clinical and commercial supplies; or
|
|
|
• |
the FDA may change its approval policies or adopt new regulations.
|
|
|
• |
regulators or IRBs may not authorize Ocuphire or its investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site including due to the ongoing COVID-19 pandemic or other public health emergency;
|
|
|
• |
government or regulatory delays and changes in regulatory requirements, policy and guidelines may require Ocuphire to perform additional clinical trials or use substantial additional resources to obtain regulatory approval, including
due to the ongoing COVID-19 pandemic or other public health emergency;
|
|
|
• |
Ocuphire may have delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites, including due to the ongoing COVID-19 pandemic or other public health
emergency;
|
|
|
• |
clinical trials may produce negative or inconclusive results, and Ocuphire may decide, or regulators may require it, to conduct additional clinical trials or abandon product development programs, including due to the ongoing COVID-19
pandemic or other public health emergency;
|
|
|
• |
the number of patients required for clinical trials may be larger, enrollment in these clinical trials may be slower or participants may drop out of these clinical trials at a higher rate than Ocuphire anticipates, including due to the
ongoing COVID-19 pandemic or other public health emergency;
|
|
|
• |
Ocuphire’s third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to Ocuphire in a timely manner, or at all;
|
|
|
• |
Ocuphire’s patients or medical investigators may be unwilling to follow its clinical trial protocols;
|
|
|
• |
Ocuphire might have to suspend or terminate clinical trials for various reasons, including a finding that the participants are being exposed to unacceptable health risks;
|
|
|
• |
the cost of clinical trials may be greater than Ocuphire anticipates, including due to the ongoing COVID-19 pandemic or other public health emergency;
|
|
|
• |
the supply or quality of any product candidate or other materials necessary to conduct clinical trials may be insufficient or inadequate;
|
|
|
• |
the product candidate may have undesirable side effects or other unexpected characteristics, causing Ocuphire or its investigators, regulators or IRBs to suspend or terminate the trials;
|
|
|
• |
clinical trials may be delayed or terminated because of the ongoing COVID-19 pandemic or another public health emergency; and
|
|
|
• |
federal agencies may, due to reduced manpower or diverted resources to the COVID-19 pandemic, require more time to review clinical trial protocols and INDs.
|
|
|
• |
severity of the disease under investigation;
|
|
|
• |
availability and efficacy of medications already approved for the disease under investigation;
|
|
|
• |
eligibility criteria for the trial in question;
|
|
|
• |
competition for eligible patients with other companies conducting clinical trials for product candidates seeking to treat the same indication or patient population;
|
|
|
• |
its payments for conducting clinical trials;
|
|
|
• |
perceived risks and benefits of the product candidate under study;
|
|
|
• |
efforts to facilitate timely enrollment in clinical trials;
|
|
|
• |
patient referral practices of physicians;
|
|
|
• |
the ability to monitor patients adequately during and after treatment;
|
|
|
• |
proximity and availability of clinical trial sites for prospective patients;
|
|
|
• |
the ability of patients to safely participate in clinical trials during the COVID-19 pandemic or other public health emergencies; and
|
|
|
• |
the ability to monitor patients adequately during periods in which social distancing is required or recommended due to the COVID-19 pandemic.
|
|
|
• |
regulatory authorities may withdraw their approval of the product;
|
|
|
• |
Ocuphire may be required to recall the product, change the way this product is administered, conduct additional clinical trials, or change the labeling or distribution of the product (including REMS);
|
|
|
• |
additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the product;
|
|
|
• |
Ocuphire may be subject to fines, injunctions, or the imposition of civil or criminal penalties;
|
|
|
• |
Ocuphire could be sued and held liable for harm caused to patients;
|
|
|
• |
the product may be rendered less competitive and sales may decrease; or
|
|
|
• |
Ocuphire’s reputation may suffer generally both among clinicians and patients.
|
|
|
• |
obtain favorable results from and complete the clinical development of both Nyxol and APX3330 for their planned indications, including successful completion of the Phase 2 and Phase 3 trials for these indications;
|
|
|
• |
submit an application to regulatory authorities for both product candidates and receive marketing approval in the United States and foreign countries;
|
|
|
• |
contract for the manufacture of commercial quantities of its product candidates at acceptable cost levels;
|
|
|
• |
establish sales and marketing capabilities to effectively market and sell its product candidates in the United States or other markets, alone or with a pharmaceutical partner; and
|
|
|
• |
achieve market acceptance of its product candidates in the medical community and with third-party payors.
|
|
|
• |
the scope, size, rate of progress, results, and costs of researching and developing its product candidates, and initiating and completing its preclinical studies and clinical trials;
|
|
|
• |
the cost, timing and outcome of its efforts to obtain marketing approval for its product candidates in the United States and other countries, including to fund the preparation and filing of an NDA with the FDA for its product
candidates and to satisfy related FDA requirements and regulatory requirements in other countries;
|
|
|
• |
the number and characteristics of any additional product candidates it develops or acquires, if any;
|
|
|
• |
Ocuphire’s ability to establish and maintain collaborations on favorable terms, if at all;
|
|
|
• |
the amount of revenue, if any, from commercial sales, should its product candidates receive marketing approval;
|
|
|
• |
the costs associated with commercializing its product candidates, if Ocuphire receives marketing approval, including the cost and timing of developing sales and marketing capabilities or entering into strategic collaborations to market
and sell its product candidates;
|
|
|
• |
the cost of manufacturing its product candidates or products Ocuphire successfully commercializes; and
|
|
|
• |
the costs associated with general corporate activities, such as the cost of filing, prosecuting and enforcing patent claims and making regulatory filings.
|
|
|
• |
litigation involving patients taking Ocuphire’s drugs;
|
|
|
• |
restrictions on such drugs, manufacturers, or manufacturing processes;
|
|
|
• |
restrictions on the labeling or marketing of a drug;
|
|
|
• |
restrictions on drug distribution or use;
|
|
|
• |
requirements to conduct post-marketing studies or clinical trials;
|
|
|
• |
warning letters or untitled letters;
|
|
|
• |
withdrawal of the drugs from the market;
|
|
|
• |
refusal to approve pending applications or supplements to approved applications that Ocuphire submits;
|
|
|
• |
product recall or public notification or medical product safety alerts to healthcare professionals;
|
|
|
• |
fines, restitution, or disgorgement of profits or revenues;
|
|
|
• |
suspension or withdrawal of marketing approvals;
|
|
|
• |
damage to relationships with any potential collaborators;
|
|
|
• |
unfavorable press coverage and damage to Ocuphire’s reputation;
|
|
|
• |
refusal to permit the import or export of drugs;
|
|
|
• |
product seizure; or
|
|
|
• |
injunctions or the imposition of civil or criminal penalties.
|
|
|
• |
the federal Anti-Kickback Statute prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or
reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
|
|
|
• |
the federal false claims and civil monetary penalties laws, including the civil False Claims Act, impose criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly
presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government;
|
|
|
• |
HIPAA imposes criminal and civil liability for, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
|
|
|
• |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and its implementing regulations, also imposes obligations, including mandatory contractual terms, on certain people and entities with respect
to safeguarding the privacy, security, and transmission of individually identifiable health information;
|
|
|
• |
the federal Physician Payments Sunshine Act under the Affordable Care Act requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s
Health Insurance Program, with specific exceptions, to report specially to the Centers for Medicare & Medicaid Services within the U.S. Department of Health and Human Services information related to physician payments and other
transfers of value and physician ownership and investment interests; and
|
|
|
• |
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental
third-party payors, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the
federal government, in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. Certain state and foreign laws also govern the privacy and
security of health information in ways that differ from each other and often are not preempted by HIPAA, thus complicating compliance efforts.
|
|
|
• |
comply with the regulations of the FDA and applicable non-U.S. regulators;
|
|
|
• |
provide accurate information to the FDA and applicable non-U.S. regulators;
|
|
|
• |
comply with healthcare fraud and abuse laws and regulations in the United States and abroad;
|
|
|
• |
report financial information or data accurately; or
|
|
|
• |
disclose unauthorized activities to Ocuphire.
|
|
|
• |
Presbysol® (AGN-190584), with 1.25% pilocarpine, developed by Allergan plc. (NDA application submitted February 2021).
|
|
|
• |
Presbidrops® (CSF-1), with low dose pilocarpine and a secondary agent (lubricant), developed by Orasis Pharmaceuticals Ltd.
|
|
|
• |
Liquid Vision®, with aceclidine (another miotic agent), developed by Presbyopia Therapies, LLC.
|
|
|
• |
MicroLine®, which is a microdose formulation of pilocarpine, developed by Eyenovia, Inc.
|
|
|
• |
KT-101, which uses pilocarpine in the AcuStream delivery system, developed by Kedalion Therapeutics, Inc.
|
|
|
• |
BrimocholTM, with brimonidine and carbachol (both are miotic agents), developed by Visus Therapeutics, Inc.
|
|
|
• |
UNR844, which uses a mechanism that involves softening the lens to increase near visual acuity, developed by Novartis AG (originally Encore Vision, Inc.).
|
|
|
• |
Lucentis® (ranibizumab) and Avastin® (bevacizumab), which are anti-VEGF monoclonal antibody intravitreal injections, developed by Genentech, Inc.
|
|
|
• |
EYLEA® (aflibercept), a VEGF inhibitor intravitreal injection, developed by Regeneron Pharmaceuticals.
|
|
|
• |
Beovu® Brolucizumab, an anti-VEGF monoclonal antibody intravitreal injection, developed by Novartis AG.
|
|
|
• |
MACUGEN® (pegaptanib sodium injection), a selective inhibitor of VEGF-165, developed by Bausch + Lomb.
|
|
|
• |
Ozurdex® (dexamethasone), a corticosteroid IVT implant, developed by Allergan plc.
|
|
|
• |
Iluvien (fluocinolone acetonide), a corticosteroid IVT implant, developed by Alimera Sciences, Inc.
|
|
|
• |
Abicipar, an anti-VEGF intravitreal injection with a long duration of action, developed by Allergan plc and Molecular Partners.
|
|
|
• |
Farcimab, a bispecific antibody intravitreal injection that suppresses both VEGF and Angiopoietin-2, developed by Genentech, Inc. and Roche AG.
|
|
|
• |
KSI-301, an anti-VEGF antibody intravitreal injection coupled with a biopolymer that is intended to increase the time between injections, developed by Kodiak Sciences.
|
|
|
• |
OPT-302, an intravitreal injection which binds to multiple types of VEGF receptors that could be used with other anti-VEGF agents, developed by Opthea Limited.
|
|
|
• |
ALG-1001, an integrin peptide therapy intravitreal injection that is being evaluated as a sequential or in-combination therapy with bevacizumab in patients with DME, developed by Allegro Ophthalmics, LLC.
|
|
|
• |
RG-7774, an orally administered selective CB2 (Cannabinoid 2) receptor agonist that is being evaluated in patients with moderately severe to severe non-proliferative diabetic retinopathy, developed by Hoffmann-LA Roche, AG.
|
|
|
• |
the inability to recruit and retain adequate numbers of effective sales and marketing personnel or enter into distribution agreements with third parties;
|
|
|
• |
the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe its product candidate;
|
|
|
• |
the lack of complementary products to be offered by sales personnel, which may put Ocuphire at a competitive disadvantage relative to companies with more extensive product lines;
|
|
|
• |
unforeseen costs and expenses associated with creating an independent sales and marketing organization; and
|
|
|
• |
the inability to obtain sufficient coverage and reimbursement from third-party payors and governmental agencies.
|
|
|
• |
efficacy and potential advantages compared to alternative treatments;
|
|
|
• |
the ability to offer Ocuphire’s product for sale at competitive prices;
|
|
|
• |
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
|
|
|
• |
any restrictions on the use of Ocuphire’s product together with other medications;
|
|
|
• |
interactions of its product with other medicines patients are taking;
|
|
|
• |
inability of certain types of patients to take Ocuphire’s product;
|
|
|
• |
demonstrated ability to treat patients and, if required by any applicable regulatory authority in connection with the approval for target indications as compared with other available therapies;
|
|
|
• |
the relative convenience and ease of administration as compared with other treatments available for approved indications;
|
|
|
• |
the prevalence and severity of any adverse side effects;
|
|
|
• |
limitations or warnings contained in the labeling approved by the FDA;
|
|
|
• |
availability of alternative treatments already approved or expected to be commercially launched in the near future;
|
|
|
• |
the effectiveness of Ocuphire’s sales and marketing strategies;
|
|
|
• |
Ocuphire’s ability to increase awareness through marketing efforts;
|
|
|
• |
guidelines and recommendations of organizations involved in research, treatment and prevention of various diseases that may advocate for alternative therapies;
|
|
|
• |
Ocuphire’s ability to obtain sufficient third-party coverage and adequate reimbursement;
|
|
|
• |
the willingness of patients to pay out-of-pocket in the absence of third-party coverage; and
|
|
|
• |
physicians or patients may be reluctant to switch from existing therapies even if potentially more effective, safe or convenient.
|
|
|
• |
decreased demand for any product candidate that Ocuphire is developing;
|
|
|
• |
injury to Ocuphire’s reputation and significant negative media attention;
|
|
|
• |
withdrawal of clinical trial participants;
|
|
|
• |
increased FDA warnings on product labels;
|
|
|
• |
significant costs to defend the related litigation;
|
|
|
• |
substantial monetary awards to trial participants or patients;
|
|
|
• |
distraction of management’s attention from Ocuphire’s primary business;
|
|
|
• |
loss of revenue; and
|
|
|
• |
the inability to commercialize any product candidate that Ocuphire may develop.
|
Ocuphire will be unable to directly control all aspects of its non-clinical studies and its clinical trials due to its reliance on CROs and other third parties that assist Ocuphire in conducting non-clinical studies and clinical trials.
|
|
• |
have staffing difficulties;
|
|
|
• |
fail to comply with contractual obligations;
|
|
|
• |
experience regulatory compliance issues;
|
|
|
• |
undergo changes in priorities or become financially distressed; or
|
|
|
• |
form relationships with other entities, some of which may be Ocuphire’s competitors.
|
|
|
• |
collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
|
|
|
• |
collaborators may not perform their obligations as expected;
|
|
|
• |
collaborators may not pursue development and commercialization or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus or available
funding, or external factors such as an acquisition that diverts resources or creates competing priorities;
|
|
|
• |
collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product
candidate for clinical testing;
|
|
|
• |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with its product candidate if the collaborators believe that competitive products are more likely to be successfully
developed or can be commercialized under terms that are more attractive than Ocuphire’s;
|
|
|
• |
a collaborator with marketing and distribution rights to one or more product candidates may not commit sufficient resources to the marketing or distribution of any such product candidate;
|
|
|
• |
collaborators may not properly maintain or defend Ocuphire’s intellectual property rights or may use its proprietary information in such a way as to invite litigation that could jeopardize or invalidate Ocuphire’s proprietary
information or expose Ocuphire to litigation;
|
|
|
• |
collaborators may infringe the intellectual property rights of third parties, which may expose Ocuphire to litigation and potential liability;
|
|
|
• |
disputes may arise between the Ocuphire and collaborators that result in the delay or termination of research, development, or commercialization of its product candidates, or in litigation or arbitration that diverts management
attention and resources;
|
|
|
• |
Ocuphire may lose certain valuable rights under circumstances identified in its collaborations, including if it undergoes a change of control;
|
|
|
• |
collaborations may be terminated and such terminations may create a need for additional capital to pursue further development or commercialization of the applicable product candidates;
|
|
|
• |
collaborators may learn about Ocuphire’s discoveries and use this knowledge to compete with Ocuphire in the future;
|
|
|
• |
the results of collaborators’ preclinical or clinical studies could harm or impair other development programs;
|
|
|
• |
there may be conflicts between different collaborators that could negatively affect those collaborations and potentially others;
|
|
|
• |
the number and nature of Ocuphire’s collaborations could adversely affect its attractiveness to potential future collaborators or acquirers;
|
|
|
• |
collaboration agreements may not lead to development or commercialization of its product candidate in the most efficient manner or at all. If a present or future collaborator of Ocuphire were to be involved in a business combination,
the continued pursuit and emphasis on its product development or commercialization program under such collaboration could be delayed, diminished, or terminated; and
|
|
|
• |
collaborators may be unable to obtain the necessary marketing approvals.
|
|
|
• |
increased operating expenses and cash requirements;
|
|
|
• |
the assumption of indebtedness or contingent liabilities;
|
|
|
• |
the issuance of Ocuphire’s equity securities which would result in dilution to Ocuphire Stockholders;
|
|
|
• |
assimilation of operations, intellectual property, products and product candidates of an acquired company, including difficulties associated with integrating new personnel;
|
|
|
• |
the diversion of management’s attention from Ocuphire’s existing product candidates and initiatives in pursuing such an acquisition or strategic partnership;
|
|
|
• |
retention of key employees, the loss of key personnel, and uncertainties in Ocuphire’s ability to maintain key business relationships;
|
|
|
• |
risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and
|
|
|
• |
Ocuphire’s inability to generate revenue from acquired intellectual property, technology and/or products sufficient to meet its objectives or even to offset the associated transaction and maintenance costs.
|
|
|
• |
any of Ocuphire’s patents, or any of its pending patent applications, if issued, will include claims having a scope sufficient to protect its product candidates;
|
|
|
• |
any of its pending patent applications will result in issued patents;
|
|
|
• |
Ocuphire will be able to successfully commercialize its product candidates, if approved, before its relevant patents expire;
|
|
|
• |
Ocuphire was the first to make the inventions covered by each of its patents and pending patent applications;
|
|
|
• |
Ocuphire was the first to file patent applications for these inventions;
|
|
|
• |
others will not develop similar or alternative technologies that do not infringe Ocuphire’s patents;
|
|
|
• |
any of Ocuphire’s patents will be valid and enforceable;
|
|
|
• |
any patents issued to Ocuphire will provide a basis for an exclusive market for its commercially viable products, will provide Ocuphire with any competitive advantages or will not be challenged by third parties;
|
|
|
• |
Ocuphire will develop additional proprietary technologies or product candidates that are separately patentable; or
|
|
|
• |
that Ocuphire’s commercial activities or products will not infringe upon the patents of others.
|
|
|
• |
the scope of rights granted under the license agreement and other interpretation-related issues;
|
|
|
• |
the extent to which Ocuphire’s product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
|
|
|
• |
the sublicensing of patent and other rights under Ocuphire’s collaborative development relationships;
|
|
|
• |
Ocuphire’s diligence obligations under the license agreement and what activities satisfy those diligence obligations;
|
|
|
• |
the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property; and
|
|
|
• |
the priority of invention of patented technology.
|
|
|
• |
compliance with differing or unexpected regulatory requirements for its product candidates;
|
|
|
• |
different medical practices and customs affecting acceptance of its product candidates, if approved, or any other approved product in the marketplace;
|
|
|
• |
language barriers;
|
|
|
• |
the interpretation of contractual provisions governed by foreign law in the event of a contract dispute;
|
|
|
• |
difficulties in staffing and managing foreign operations, and an inability to control commercial or other activities where it is relying on third parties;
|
|
|
• |
workforce uncertainty in countries where labor unrest is more common than in the United States;
|
|
|
• |
potential liability under the Foreign Corrupt Practice Act of 1977 or comparable foreign regulations;
|
|
|
• |
production shortages resulting from any events affecting raw material supply or manufacturing capability abroad;
|
|
|
• |
foreign government taxes, regulations, and permit requirements;
|
|
|
• |
U.S. and foreign government tariffs, trade restrictions, price and exchange controls, and other regulatory requirements;
|
|
|
• |
economic weakness, including inflation, natural disasters, war, events of terrorism, or political instability in particular foreign countries;
|
|
|
• |
fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenues;
|
|
|
• |
compliance with tax, employment, immigration, and labor laws, regulations, and restrictions for employees living or traveling abroad;
|
|
|
• |
changes in diplomatic and trade relationships; and
|
|
|
• |
challenges in enforcing its contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States.
|
|
|
• |
interruption in global manufacturing and shipping that has affected, and may continue to affect the transport of clinical trial materials and materials, including testing equipment and personal protective equipment;
|
|
|
• |
changes in local regulations as part of a response to the COVID-19 coronavirus outbreak which may result in unexpected costs;
|
|
|
• |
delay in the timing of interactions with the FDA due to absenteeism by federal employees or by the diversion of their efforts and attention to approval of other therapeutics or other activities related to COVID-19;
|
|
|
• |
impacts on Ocuphire’s ability to secure additional financing on favorable terms; and
|
|
|
• |
modifications to the Ocuphire convertible notes.
|
|
|
• |
the announcement of new products or product enhancements by Ocuphire or its competitors;
|
|
|
• |
changes in Ocuphire's relationships with its licensors or other strategic partners;
|
|
|
• |
developments concerning intellectual property rights and regulatory approvals;
|
|
|
• |
variations in Ocuphire's and Ocuphire's competitors’ results of operations;
|
|
|
• |
substantial sales of shares of our common stock due to the release of lock-up agreements;
|
|
|
• |
the announcement of clinical trial results;
|
|
|
• |
the announcement of potentially dilutive financings;
|
|
|
• |
changes in earnings estimates or recommendations by securities analysts;
|
|
|
• |
changes in the structure of healthcare payment systems; and
|
|
|
• |
developments and market conditions in the pharmaceutical and biotechnology industries, including due to the COVID-19 pandemic.
|
| ITEM 1B. |
| ITEM 2. |
| ITEM 3. |
| ITEM 4. |
| ITEM 5. |
| ITEM 6. |
| ITEM 7. |
|
|
• |
continues clinical trials for Nyxol, APX3330 and for any other product candidate in its future pipeline;
|
|
|
• |
continues preclinical studies for Nyxol, APX3330 and for any other product candidate in its future pipeline;
|
|
|
• |
develops additional product candidates that it identifies, in-licenses or acquires;
|
|
|
• |
seeks regulatory approvals for any product candidates that successfully complete clinical trials;
|
|
|
• |
contracts to manufacture its product candidates;
|
|
|
• |
establishes on its own or with partners, a sales, marketing and distribution infrastructure to commercialize any products for which Ocuphire may obtain regulatory approval;
|
|
|
• |
maintains, expands and protects its intellectual property portfolio;
|
|
|
• |
hires additional staff, including clinical, scientific, operational and financial personnel, to execute its business plan;
|
|
|
• |
adds operational, financial and management information systems and personnel, including personnel to support its product development and potential future commercialization efforts; and
|
|
|
• |
operates as a public company.
|
|
|
• |
per patient trial costs;
|
|
|
• |
the number of patients that participate in the trials;
|
|
|
• |
the number of sites included in the trials;
|
|
|
• |
the countries in which the trials are conducted;
|
|
|
• |
the length of time required to enroll eligible patients;
|
|
|
• |
the number of doses that patients receive;
|
|
|
• |
the drop-out or discontinuation rates of patients;
|
|
|
• |
potential additional safety monitoring or other studies requested by regulatory agencies;
|
|
|
• |
the duration of patient follow-up;
|
|
|
• |
the phase of development of the product candidate;
|
|
|
• |
arrangements with contract research organizations and other service providers; and
|
|
|
• |
the efficacy and safety profile of the product candidates.
|
|
For the Year Ended
December 31,
|
||||||||||||
|
2020
|
2019
|
Change
|
||||||||||
|
Operating expenses:
|
||||||||||||
|
General and administrative
|
$
|
2,818
|
$
|
1,820
|
$
|
998
|
||||||
|
Research and development
|
6,648
|
2,373
|
4,275
|
|||||||||
|
Acquired in‑process research and development
|
10,502
|
—
|
10,502
|
|||||||||
|
Total operating expenses
|
19,968
|
4,193
|
15,775
|
|||||||||
|
Loss from operations
|
(19,968
|
)
|
(4,193
|
)
|
(15,775
|
)
|
||||||
|
Interest expense
|
(6,847
|
)
|
(1,409
|
)
|
(5,438
|
)
|
||||||
|
Fair value change in derivative and warrant liabilities
|
(1,486
|
)
|
(499
|
)
|
(987
|
)
|
||||||
|
Gain on note extinguishment
|
3,672
|
—
|
3,672
|
|||||||||
|
Other income (expense), net
|
9
|
(68
|
)
|
77
|
||||||||
|
Loss before income taxes
|
(24,620
|
)
|
(6,169
|
)
|
(18,451
|
)
|
||||||
|
Provision for income taxes
|
—
|
—
|
—
|
|||||||||
|
Net loss
|
$
|
(24,620
|
)
|
$
|
(6,169
|
)
|
$
|
(18,451
|
)
|
|||
|
|
• |
continues clinical trials and preclinical studies for Nyxol, APX 3330 and for any other product candidate in its future pipeline;
|
|
|
• |
develops additional product candidates that it identifies, in-licenses or acquires;
|
|
|
• |
seeks regulatory approvals for any product candidates that successfully complete clinical trials;
|
|
|
• |
contracts to manufacture its product candidates;
|
|
|
• |
establishes on its own or with partners, a sales, marketing and distribution infrastructure to commercialize any products for which it may obtain regulatory approval;
|
|
|
• |
maintains, expands and protects its intellectual property portfolio;
|
|
|
• |
hires additional staff, including clinical, scientific, operational and financial personnel, to execute its business plan;
|
|
|
• |
adds operational, financial and management information systems and personnel, including personnel to support its product development and potential future commercialization efforts; and
|
|
|
• |
operates as a public company.
|
|
|
• |
Qualified Financing or IPO: An amount of shares of Ocuphire common stock equal to 135% of the Note Value divided by the per share price of Ocuphire
common stock issued to purchasers in the Qualified Financing or IPO.
|
|
|
• |
CIC: An amount of shares of Ocuphire common stock equal to 200% of the Note Value divided by the per share price of Ocuphire common stock based on the
valuation of such CIC.
|
|
|
• |
Reverse Merger: Either (i) shares of Ocuphire common stock issued in the Reverse Merger or (ii) equity securities of the Reverse Merger counterparty, in an amount equal to 135% of the Note Value
divided by the per share price at which such shares were issued to either stockholders of Ocuphire or stockholders of the Reverse Merger counterparty.
|
|
|
• |
IPO: An amount of shares of Ocuphire common stock equal to the greater of: (i) 150% of the Note Value divided by the per share price of Ocuphire common
stock issued to purchasers in the IPO, and (ii) 100% of the Note Value divided by the per share price of $10.37.
|
|
|
• |
CIC: An amount of shares of Ocuphire common stock equal to the greater of: (i) 200% of the Note Value divided by the per share price of Ocuphire common
stock based on the valuation of such CIC, and (ii) 100% of the Note Value divided by the per share price of $10.37.
|
|
|
• |
Qualified Financing: An amount of shares of Ocuphire common stock equal to 150% of the Note Value divided by the per share price of Ocuphire common
stock issued to purchasers in the Qualified Financing.
|
|
|
• |
Reverse Merger: Either shares of Ocuphire common stock issued in the Reverse Merger or equity securities of the Reverse Merger counterparty, in an amount equal to the greater of: (i) 150% of the
Note Value divided by the per share price at which such shares were issued to either stockholders of Ocuphire or stockholders of the Reverse Merger counterparty, and (ii) 100% Note Value divided by the per share price of $10.37.
|
|
For the Year Ended
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Net cash used in operating activities
|
$
|
(6,797
|
)
|
$
|
(3,593
|
)
|
||
|
Net cash provided by (used in) investing activities
|
539
|
(25
|
)
|
|||||
|
Net cash provided by financing activities
|
21,120
|
4,704
|
||||||
|
Net increase in cash and cash equivalents
|
$
|
14,862
|
$
|
1,086
|
||||
| ITEM 7A. |
| ITEM 8. |
| ITEM 9. |
| ITEM 9A. |
| ITEM 9B. |
| ITEM 10. |
| ITEM 11. |
| ITEM 12. |
| ITEM 13. |
| ITEM 14. |
| ITEM 15. |
|
EXHIBIT
|
||
|
NUMBER
|
|
DESCRIPTION OF DOCUMENT
|
|
|
Agreement and Plan of Merger, dated as of June 17, 2020, by and among the Registrant, Razor Merger Sub, Inc. and Ocuphire Pharma, Inc. (incorporated by reference to Exhibit 2.1 to the Registrant’s Current
Report on Form 8-K, filed on June 19, 2020)..
|
|
|
|
First Amendment to Agreement and Plan of Merger and Reorganization, dated as of June 29, 2020, by and among Rexahn, Merger Sub and Ocuphire (incorporated by reference to Exhibit 2.1 to the Registrant’s Current
Report on Form 8-K , filed on July 1, 2020).
|
|
|
|
Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Appendix G to the Registrant’s Definitive Proxy Statement on Schedule 14A, filed on April 29, 2005).
|
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed on May 5, 2017).
|
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed on August 30,
2018).
|
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed on April 12,
2019).
|
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2020).
|
|
|
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.2 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2020).
|
|
|
|
Second Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.3 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2020).
|
|
|
|
Specimen Certificate for the Registrant’s Common Stock, par value $.0001 per share (incorporated by reference to Exhibit 4.3 to the Registrant’s Registration Statement on Form S-8 (File No. 333-129294), filed
on October 28, 2005).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2015).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on February 26, 2016).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on September 14, 2016).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on June 7, 2017).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on October 13, 2017).
|
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on October 19, 2018).
|
|
|
Form of Common Stock Purchase Warrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on January 25, 2019).
|
|
|
|
Stockholders Agreement, dated as of April 10, 2018, among Ocuphire Pharma, Inc. and Stockholders as defined therein (incorporated by reference to Exhibit 4.9 to the Registrant’s Registration Statement on Form
S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Form of Series A/B Warrants (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K, filed on July 1, 2020).
|
|
|
|
Description of Securities.
|
|
|
|
Rexahn Pharmaceuticals, Inc. Stock Option Plan, as amended (incorporated by reference to Exhibit 4.4 to the Registrant’s Registration Statement on Form S-8 (File No. 333-129294), filed on October 28, 2005).
|
|
|
|
Form of Stock Option Grant Agreement for Employees (incorporated by reference to Exhibit 4.5.1 to the Registrant’s Registration Statement on Form S-8 (File No. 333-129294), filed on October 28, 2005).
|
|
|
|
Form of Stock Option Grant Agreement for Non-Employee Directors and Consultants (incorporated by reference to Exhibit 4.5.2 to the Registrant’s Registration Statement on Form S-8 (File No. 333-129294), filed
on October 28, 2005).
|
|
|
|
Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan, as amended and restated (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed on June 10, 2016)
|
|
|
|
First Amendment to the Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan, as amended and restated as of June 9, 2016 (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K,
filed on April 13, 2017).
|
|
|
|
Form of Stock Option Grant Agreement under the Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan (incorporated by reference to Exhibit 10.5 to the Registrant’s Annual Report on Form 10-K for the year ended
December 31, 2015).
|
|
|
|
Form of Restricted Stock Unit Agreement under the Rexahn Pharmaceuticals, Inc. 2013 Stock Option Plan (incorporated by reference to Exhibit 10.7 to the Registrant’s Annual Report on Form 10-K for the year
ended December 31, 2018).
|
|
|
|
Lease Agreement, dated June 5, 2009, by and between Rexahn Pharmaceuticals, Inc. and The Realty Associates Fund V, L.P. (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form
10-Q for the quarterly period ended June 30, 2009).
|
|
|
|
First Amendment to Lease Agreement, dated as of June 7, 2013, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended June 30, 2013).
|
|
|
|
Second Amendment to Lease Agreement, dated as of July 26, 2014, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended September 30, 2014).
|
|
|
|
Third Amendment to Lease Agreement, dated as of May 6, 2015, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended June 30, 2015).
|
|
|
|
Fourth Amendment to Lease Agreement, dated as of April 4, 2016, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended June 30, 2016).
|
|
|
Fifth Amendment to Lease Agreement, dated as of April 13, 2017, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended June 30, 2017).
|
|
|
|
Sixth Amendment to Lease Agreement, dated as of March 19, 2019, by and between Rexahn Pharmaceuticals, Inc. and SG Plaza Holdings, LLC (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly
Report on Form 10-Q for the quarterly period ended March 31, 2019).
|
|
|
|
Form of Securities Purchase Agreement, dated as of November 6, 2015, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on November 6, 2015).
|
|
|
|
Form of Securities Purchase Agreement, dated as of February 26, 2016, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on February 26, 2016).
|
|
|
|
Form of Securities Purchase Agreement, dated as of September 14, 2016, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on September 14, 2016).
|
|
|
|
Form of Securities Purchase Agreement, dated as of June 6, 2017, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on June 7, 2017).
|
|
|
|
Form of Securities Purchase Agreement, dated as of October 13, 2017, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on October 13, 2017).
|
|
|
|
Form of Securities Purchase Agreement, dated as of October 17, 2018, by and between the Company and the purchasers identified on the signature pages thereto (incorporated by reference to Exhibit 10.1 to the
Registrant’s Current Report on Form 8-K, filed on October 19, 2018).
|
|
|
|
Collaboration and License Agreement, dated as of February 25, 2019, between BioSense Global, LLC and the Company (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q for
the quarterly period ended June 30, 2019).
|
|
|
|
Amendment No. 1 to Collaboration and License Agreement, dated as of August 24, 2019, between BioSense Global LLC and the Company (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report
on Form 8-K filed on August 29, 2019).
|
|
|
|
Amendment No. 2 to Collaboration and License Agreement, dated as of March 10, 2020 between BioSense Global LLC and the Company (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report
on Form 10-Q filed on May 7, 2020).
|
|
|
|
Amended and Restated Employment Agreement by and among the Company and Mina Sooch, effective as of November 5, 2020 (incorporated by reference to Exhibit 10.27 to the Registrant’s Registration Statement on
Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Amended and Restated Employment Agreement by and among the Company and Bernhard Hoffmann, effective as of November 5, 2020 (incorporated by reference to Exhibit 10.29 to the Registrant’s Registration
Statement on Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Form of Indemnification Agreement (incorporated by reference to Exhibit 10.30 to the Registrant’s Registration Statement on Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Sublicense Agreement, dated as of January 21, 2020, by and between Ocuphire Pharma, Inc. and Apexian Pharmaceuticals, Inc (incorporated by reference to Exhibit 10.31 to the Registrant’s Registration Statement
on Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
First Amendment to Sublicense Agreement, dated as of June 4, 2020, by and between Apexian Pharmaceuticals, Inc. and Ocuphire Pharma, Inc (incorporated by reference to Exhibit 10.32 to the Registrant’s
Registration Statement on Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Lease Agreement, dated as of May 19, 2019, by and between Ocuphire Pharma, Inc. and Duke & Duke, LP (incorporated by reference to Exhibit 10.33 to the Registrant’s Registration Statement on Form S-4
(File No. 333-239702), filed on September 30, 2020).
|
|
|
|
First Amendment to Lease Agreement, dated as of October 29, 2019, by and between Ocuphire Pharma, Inc. and Duke & Duke, LP (incorporated by reference to Exhibit 10.34 to the Registrant’s Registration
Statement on Form S-4 (File No. 333-239702), filed on September 30, 2020).
|
|
|
|
Ocuphire Pharma, Inc. 2018 Equity Incentive Plan, dated as of April 9, 2019 (incorporated by reference to Exhibit 10.35 to the Registrant’s Registration Statement on Form S-4 (File No. 333-239702), filed on
September 30, 2020).
|
|
|
|
First Amendment to 2018 Equity Incentive Plan, dated as of December 23, 2019 (incorporated by reference to Exhibit 10.36 to the Registrant’s Registration Statement on Form S-4 (File No. 333-239702), filed on
September 30, 2020).
|
|
|
|
Form of Option Agreement issuable under the Ocuphire Pharma, Inc. 2018 Equity Incentive Plan (incorporated by reference to Exhibit 10.37 to the Registrant’s Registration Statement on Form S-4 (File No.
333-239702), filed on September 30, 2020).
|
|
|
|
Ocuphire Pharma, Inc. 2020 Omnibus Equity Incentive Plan (incorporated by reference to Exhibit 10.38 to the Registrant’s Registration Statement on Form S-4 (File No. 333-239702), filed on September 30,
2020).
|
|
|
|
Amended and Restated Securities Purchase Agreement, dated as of June 29, 2020, by and among Rexahn, Ocuphire and the investors party thereto (incorporated by reference to Exhibit 10.1 to the Registrant’s
Current Report on Form 8-K, filed on July 1, 2020).
|
|
|
|
Form of Waiver Agreement, dated as of February 3, 2021, by and between Ocuphire Pharma, Inc. and the Holder(s) (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed
on February 4, 2021)
|
|
|
|
Form of Financing Lock-Up Agreement, by and among the Company, OcuSub, Inc., and the investors party thereto (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K, filed on
July 1, 2020).
|
|
|
|
Form of Lock-Up Agreement, by and between the Company, OcuSub, Inc. and certain stockholders
|
|
|
|
Form of Leak-Out Agreement, by and between the Company and the investors party thereto (incorporated by reference to Exhibit 10.3 to the Registrant’s Current Report on Form 8-K, filed on July 1, 2020).
|
|
|
|
Contingent Value Rights Agreement, dated as of November 5, 2020, by and among the Company, Shareholder Representative Services LLC and the Olde Monmouth Stock Transfer Co., Inc. (incorporated by reference
to Exhibit 10.4 to the Registrant’s Current Report on Form 8-K, filed on November 6, 2020).
|
|
|
|
Ocuphire Pharma, Inc. 2020 Inducement Plan
|
|
|
|
Employment Agreement dated November 11, 2020, by and between the Company and Amy Rabourn
|
|
|
10.43 |
Second Lease Amendment, dated as of November 17, 2020, by and between the Company and Duke & Duke
|
|
|
|
Subsidiaries of the Registrant
|
|
|
|
Consent of Ernst & Young, LLP.
|
|
|
|
Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of
2002
|
|
|
|
Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as adopted pursuant to section 302 of the Sarbanes-Oxley Act of
2002
|
|
|
|
Certification of Principal Executive Officer and Principal Financial Officer pursuant to Rules 13a-14(b) and 15d-14(b) promulgated under the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350, as
adopted pursuant to section 906 of The Sarbanes-Oxley Act of 2002
|
|
101.INS
|
|
XBRL Instance Document
|
|
101.SCH
|
|
XBRL Taxonomy Extension Schema Document
|
|
101.CAL
|
|
XBRL Taxonomy Extension Calculation Linkbase Document
|
|
101.DEF
|
|
XBRL Taxonomy Extension Definition Linkbase Document
|
|
101.LAB
|
|
XBRL Taxonomy Extension Label Linkbase Document
|
|
101.PRE
|
|
XBRL Taxonomy Extension Presentation Linkbase Document
|
|
*
|
Indicates management contract or compensatory plan.
|
|
+
|
Certain schedules and exhibits have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request.
|
|
++
|
Portions of this exhibit have been omitted in compliance with Item 601 of Regulation S-K.
|
| ITEM 16. |
| ITEM 8. |
| 133 | |
| 135 |
|
|
136
|
|
| 137 |
|
| 138 |
|
| 139 |
|
|
Accounting for Warrants
|
|
|
Description of
the Matter
|
As discussed in Notes 1 and 9 to the consolidated financial statements, in connection with the merger transaction, the Company issued Series A and Series B warrants (collectively, the Warrants) to purchase additional common stock on
November 19, 2020 (the Warrant Closing Date). As of the Warrant Closing Date, the Series A warrants were determined to be liability classified instruments and the Series B warrants were determined to be equity classified instruments.
|
|
|
Auditing the accounting conclusions for the issuance of the Warrants was challenging because of the complex provisions affecting valuation and classification and required extensive audit effort. The accounting for the issuance of the
Warrants involved an assessment of the particular features of each type of warrant, and the impact of those features on the accounting and classification of the Warrants.
|
||
|
How we
Addressed the
Matter in Our
Audit
|
To test the accounting and determine proper classification of the Warrants, our audit procedures included, among others, inspecting the agreements and evaluating the completeness and accuracy of the Company’s technical accounting
analyses, application of the relevant accounting guidance and review of legal interpretation from counsel. Our audit procedures also included the involvement of subject matter resources to assist in evaluating management’s conclusion on
the interpretation and application of the relevant accounting literature.
|
|
As of December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Assets
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$
|
16,399
|
$
|
1,537
|
||||
|
Prepaids and other assets
|
1,269
|
149
|
||||||
|
Deferred costs
|
—
|
76
|
||||||
|
Total current assets
|
17,668
|
1,762
|
||||||
|
Property and equipment, net
|
14
|
22
|
||||||
|
Total assets
|
$
|
17,682
|
$
|
1,784
|
||||
|
Liabilities and stockholders’ deficit
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$
|
1,214
|
$
|
342
|
||||
|
Accrued expenses
|
1,971
|
621
|
||||||
|
Convertible notes
|
—
|
4,977
|
||||||
|
Convertible notes from related parties
|
—
|
690
|
||||||
|
Premium conversion derivatives
|
—
|
2,714
|
||||||
|
Total current liabilities
|
3,185
|
9,344
|
||||||
|
Warrant liabilities
|
27,964
|
—
|
||||||
|
Total liabilities
|
31,149
|
9,344
|
||||||
|
Commitments and contingencies (Note 3 and Note 9)
|
||||||||
|
Stockholders’ deficit
|
||||||||
|
Preferred stock, par value $0.0001; 10,000,000 and 625,000 shares authorized as of December 31, 2020 and 2019, respectively; no shares issued and outstanding at December 31, 2020 and
2019.
|
—
|
—
|
||||||
|
Common stock, par value $0.0001; 75,000,000 and 5,000,000 shares authorized as of December 31, 2020 and 2019, respectively; 10,882,495 and 2,852,485 shares issued and outstanding at
December 31, 2020 and 2019, respectively.
|
1
|
—
|
||||||
|
Additional paid-in capital
|
19,207
|
495
|
||||||
|
Accumulated deficit
|
(32,675
|
)
|
(8,055
|
)
|
||||
|
Total stockholders’ deficit
|
(13,467
|
)
|
(7,560
|
)
|
||||
|
Total liabilities and stockholders’ deficit
|
$
|
17,682
|
$
|
1,784
|
||||
|
For the Year Ended
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Operating expenses:
|
||||||||
|
General and administrative
|
$
|
2,818
|
$
|
1,820
|
||||
|
Research and development
|
6,648
|
2,373
|
||||||
|
Acquired in-process research and development
|
10,502
|
—
|
||||||
|
Total operating expenses
|
19,968
|
4,193
|
||||||
|
Loss from operations
|
(19,968
|
)
|
(4,193
|
)
|
||||
|
Interest expense
|
(6,847
|
)
|
(1,409
|
)
|
||||
|
Fair value change in derivative and warrant liabilities
|
(1,486
|
)
|
(499
|
)
|
||||
|
Gain on note extinguishment
|
3,672
|
—
|
||||||
|
Other income (expense), net
|
9
|
( 68
|
)
|
|||||
|
Loss before income taxes
|
(24,620
|
)
|
(6,169
|
)
|
||||
|
Benefit (provision) for income taxes
|
—
|
—
|
||||||
|
Net loss
|
(24,620
|
)
|
(6,169
|
)
|
||||
|
Other comprehensive loss, net of tax
|
—
|
—
|
||||||
|
Comprehensive loss
|
$
|
(24,620
|
)
|
$
|
(6,169
|
)
|
||
|
Net loss per share:
|
||||||||
|
Basic and diluted (Note 10)
|
$
|
(5.28
|
)
|
$
|
(2.17
|
)
|
||
|
Number of shares used in per share calculations:
|
||||||||
|
Basic and diluted
|
4,661,110
|
2,844,832
|
||||||
|
Common Stock
|
Additional
Paid–In
|
Accumulated
|
Total
Stockholders’
|
|||||||||||||||||
|
Shares
|
Amount
|
Capital
|
Deficit
|
Deficit
|
||||||||||||||||
|
Balance at December 31, 2018
|
2,852,485
|
$
|
—
|
$
|
187
|
$
|
(1,886
|
)
|
$
|
(1,699
|
)
|
|||||||||
|
Share-based compensation
|
—
|
—
|
308
|
—
|
308
|
|||||||||||||||
|
Net and comprehensive loss
|
—
|
—
|
—
|
(6,169
|
)
|
(6,169
|
)
|
|||||||||||||
|
Balance at December 31, 2019
|
2,852,485
|
—
|
495
|
(8,055
|
)
|
(7,560
|
)
|
|||||||||||||
|
Issuance of common stock in exchange for in-process research and development
|
891,422
|
—
|
2,126
|
—
|
2,126
|
|||||||||||||||
|
Gain on note extinguishment
|
—
|
—
|
971
|
—
|
971
|
|||||||||||||||
|
Conversion of convertible notes into common stock upon close of the merger
|
977,128
|
—
|
6,953
|
—
|
6,953
|
|||||||||||||||
|
Issuance of common stock and warrants in connection with pre-merger financing
|
4,999,988
|
1
|
(1
|
)
|
—
|
—
|
||||||||||||||
|
Issuance costs attributed to pre-merger financing
|
—
|
—
|
(1,080
|
)
|
—
|
(1,080
|
)
|
|||||||||||||
|
Issuance of common stock, warrants and options to former Rexahn stockholders and effect of asset acquisition
|
1,120,800
|
—
|
8,115
|
—
|
8,115
|
|||||||||||||||
|
Share–based compensation
|
—
|
—
|
1,506
|
—
|
1,506
|
|||||||||||||||
|
Reclassification of Rexahn warrants from liability to equity
|
—
|
—
|
64
|
—
|
64
|
|||||||||||||||
|
Exercise of stock options
|
40,672
|
—
|
58
|
—
|
58
|
|||||||||||||||
|
Net and comprehensive loss
|
—
|
—
|
—
|
(24,620
|
)
|
(24,620
|
)
|
|||||||||||||
|
Balance at December 31, 2020
|
10,882,495
|
$
|
1
|
$
|
19,207
|
$
|
(32,675
|
)
|
$
|
(13,467
|
)
|
|||||||||
|
For the Year Ended
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Operating activities
|
||||||||
|
Net loss
|
$
|
(24,620
|
)
|
$
|
(6,169
|
)
|
||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||
|
Share-based compensation
|
1,506
|
308
|
||||||
|
Depreciation
|
8
|
3
|
||||||
|
Non-cash acquired in-process research and development
|
10,502
|
—
|
||||||
|
Gain on note extinguishment
|
(3,672
|
)
|
—
|
|||||
|
Non-cash interest on convertible notes
|
492
|
252
|
||||||
|
Non-cash interest on convertible notes – related party
|
51
|
42
|
||||||
|
Non-cash discount amortization on convertible notes
|
873
|
1,014
|
||||||
|
Non-cash discount amortization on convertible notes – related party
|
71
|
101
|
||||||
|
1,486
|
499
|
|||||||
|
Non-cash interest attributed to Series A warrant issuance
|
4,671
|
—
|
||||||
|
Issuance costs attributed to Series A warrants
|
689
|
—
|
||||||
|
Change in assets and liabilities:
|
||||||||
|
Prepaid expenses and other assets
|
(906
|
)
|
58
|
|||||
|
Accounts payable
|
792
|
(168
|
)
|
|||||
|
Accrued and other liabilities
|
1,260
|
467
|
||||||
|
Net cash used in operating activities
|
(6,797
|
)
|
(3,593
|
)
|
||||
|
Investing activities
|
||||||||
|
Cash acquired in connection with asset acquisition
|
2,014
|
—
|
||||||
|
Transaction costs in connection with asset acquisition
|
(1,475
|
)
|
—
|
|||||
|
Purchases of property and equipment
|
—
|
(25
|
)
|
|||||
|
Net cash provided by (used in) investing activities
|
539
|
(25
|
)
|
|||||
|
Financing activities
|
||||||||
|
Proceeds from pre-merger financing
|
21,150
|
—
|
||||||
|
Proceeds from issuance of convertible notes
|
2,197
|
4,383
|
||||||
|
Proceeds from issuance of convertible notes – related party
|
—
|
323
|
||||||
|
Issuance costs attributed to pre-merger financing
|
(1,769
|
)
|
—
|
|||||
|
Issuance costs attributed to convertible notes
|
(10
|
)
|
(2
|
)
|
||||
|
Settlement of Rexahn warrants
|
(506
|
)
|
—
|
|||||
|
Exercise of stock options
|
58
|
—
|
||||||
|
Net cash provided by financing activities
|
21,120
|
4,704
|
||||||
|
Net increase in cash and cash equivalents
|
14,862
|
1,086
|
||||||
|
Cash and cash equivalents at beginning of period
|
1,537
|
451
|
||||||
|
Cash and cash equivalents at end of period
|
$
|
16,399
|
$
|
1,537
|
||||
|
Supplemental disclosure of cash flow information:
|
||||||||
|
Cash paid for income taxes
|
$
|
—
|
$
|
—
|
||||
|
Cash paid for interest
|
$
|
—
|
$
|
—
|
||||
|
Supplemental non-cash financing transactions:
|
||||||||
|
Non-cash conversion of convertible notes to common stock
|
$
|
9,365
|
$
|
—
|
||||
|
Common stock and warrants issued in connection with the asset acquisition
|
$
|
8,883
|
$
|
—
|
||||
|
Unpaid transaction costs in connection with asset acquisition
|
$
|
100
|
$
|
—
|
||||
|
Net assets assumed in connection with asset acquisition
|
$
|
68
|
$
|
—
|
||||
|
Bifurcation and modification of premium conversion derivative related to convertible notes
|
$
|
831
|
$
|
1,910
|
||||
|
Unpaid deferred offering costs
|
$
|
—
|
$
|
76
|
||||
|
Proceeds receivable from convertible note issuance
|
$
|
—
|
$
|
125
|
||||
| 1. |
Company Description and Summary of Significant Accounting Policies
|
|
|
• |
Level 1 inputs: Unadjusted quoted prices for identical assets or liabilities in active markets;
|
|
|
• |
Level 2 inputs: Quoted prices for similar assets and liabilities in active markets, quoted prices in markets that are not active, or inputs which are observable, whether directly or indirectly, for substantially the full term of the
asset or liability; and
|
|
|
• |
Level 3 inputs: Unobservable inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability in which there is little, if any, market activity for the asset or
liability at the measurement date.
|
|
As of December 31, 2020
|
||||||||||||||||
|
Description
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrant liabilities
|
$
|
27,964
|
$
|
—
|
$
|
—
|
$
|
27,964
|
||||||||
|
Total liabilities at fair value
|
$
|
27,964
|
$
|
—
|
$
|
—
|
$
|
27,964
|
||||||||
|
As of December 31, 2019
|
||||||||||||||||
|
Description
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Liabilities:
|
||||||||||||||||
|
Premium conversion derivatives
|
$
|
2,714
|
$
|
—
|
$
|
—
|
$
|
2,714
|
||||||||
|
Total liabilities at fair value
|
$
|
2,714
|
$
|
—
|
$
|
—
|
$
|
2,714
|
||||||||
|
2020
|
||||
|
Warrant liabilities
|
||||
|
Balance as of beginning of period
|
$
|
—
|
||
|
Value assigned to warrants upon in connection with pre-merger financing
|
25,821
|
|||
|
Issuance of warrants to former Rexahn stockholders classified as a liability
|
768
|
|||
|
Cash settlement of warrant liabilities
|
(506
|
)
|
||
|
Reclassification of Rexahn warrants from liability to equity
|
(64
|
)
|
||
|
Change in fair value of warrant liability
|
1,945
|
|||
|
Balance as of end of period
|
$
|
27,964
|
||
|
2020
|
2019
|
|||||||
|
Premium conversion derivatives
|
||||||||
|
Balance as of beginning of period
|
$
|
2,714
|
$
|
305
|
||||
|
Value assigned to the underlying derivatives in connection with convertible notes
|
831
|
1,910
|
||||||
|
Revaluation due to convertible note extinguishment
|
(3,086
|
)
|
—
|
|||||
|
Change in fair value of premium conversion derivatives
|
(459
|
)
|
499
|
|||||
|
Balance as of end of period
|
$
|
—
|
$
|
2,714
|
||||
| 2. |
Merger
|
|
|
• |
90% of all payments received by Rexahn or its affiliates during such CVR Payment Period from or on behalf of BioSense Global LLC (“BioSense”) pursuant to that certain License and Assignment Agreement, dated
as of February 25, 2019, by and between BioSense and Rexahn, as amended by Amendment No. 1, dated August 24, 2019, and as further amended by Amendment No. 2, dated March 10, 2020, minus certain permitted deductions;
|
|
|
• |
90% of all payments received by Rexahn or its affiliates during such CVR Payment Period from or on behalf of Zhejiang HaiChang Biotechnology Co., Ltd. (“HaiChang”) pursuant to that certain Exclusive License
Agreement, dated as of February 8, 2020, by and between HaiChang and Rexahn, minus certain permitted deductions; and
|
|
|
• |
75% of the sum of (i) all cash consideration paid by a third party to Rexahn or its affiliates during the applicable CVR Payment Period in connection with the grant, sale or transfer of rights to Rexahn’s
pre-Closing intellectual property (other than a grant, sale or transfer of rights involving a sale or disposition of the post-Merger combined company) that is entered into during the 10-year period after the Closing (“Parent IP Deal”),
plus (ii) with respect to any non-cash consideration received by Rexahn or its affiliates from a third party during the applicable CVR Payment Period in connection with any Parent IP Deal, all amounts received by Rexahn and its affiliates
for such non-cash consideration at the time such non-cash consideration is monetized by Rexahn or its affiliates, minus (iii) certain permitted deductions.
|
|
Number of shares of the combined organization owned by the Company’s Pre-Merger stockholders
|
1,120,800
|
|||
|
Multiplied by the fair value per share of REXN’s common stock (1)
|
$
|
7.24
|
||
|
Fair value of common stock issued to affect the Merger
|
8,115
|
|||
|
Fair value of warrants and options issued to affect the Merger
|
768
|
|||
|
Transaction costs
|
1,575
|
|||
|
Purchase price
|
$
|
10,458
|
|
|
(1) |
Based on the last reported sale price of the Rexahn’s common stock on the Nasdaq Capital Market on November 5, 2020, the closing date of the Merger, and gives effect to the Reverse Stock Split.
|
|
Cash acquired
|
$
|
2,014
|
||
|
Net assets assumed
|
68
|
|||
|
IPR&D (2)
|
8,376
|
|||
|
Purchase price
|
$
|
10,458
|
|
|
(2) |
Represents the pre-Merger research and development projects of Rexahn which were in-process, but not yet completed, and which the Company may advance post-Merger. This includes the development of RX-3117, RX-0301 and RX-0047. Current
accounting standards require that the fair value of IPR&D projects acquired in an asset acquisition with no alternative future use be allocated a portion of the consideration transferred and charged to expense on the acquisition date.
The acquired assets did not have outputs or employees.
|
| 3. |
Commitments and Contingencies
|
| 4. |
Supplemental Balance Sheet Information
|
|
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Prepaids
|
$
|
1,243
|
$
|
19
|
||||
|
Proceeds receivable from convertible note financing
|
—
|
75
|
||||||
|
Proceeds receivable from convertible note financing – related party
|
—
|
50
|
||||||
|
Other
|
26
|
5
|
||||||
|
Total prepaids and other assets
|
$
|
1,269
|
$
|
149
|
||||
|
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
Equipment
|
$
|
20
|
$
|
20
|
||||
|
Furniture
|
5
|
5
|
||||||
|
Total property and equipment
|
25
|
25
|
||||||
|
Less accumulated depreciation
|
(11
|
)
|
(3
|
)
|
||||
|
Property and equipment, net
|
$
|
14
|
$
|
22
|
||||
|
Accrued expenses consist of the following (in thousands):
|
December 31,
|
|||||||
|
2020
|
2019
|
|||||||
|
R&D services and supplies
|
$
|
1,440
|
$
|
—
|
||||
|
Payroll
|
320
|
350
|
||||||
|
Professional services
|
186
|
262
|
||||||
|
Other
|
25
|
9
|
||||||
|
Total
|
$
|
1,971
|
$
|
621
|
||||
| 5. |
Convertible Notes
|
|
|
• |
IPO: The Convertible Notes would have automatically converted into the number of fully paid and non-assessable shares of the Company’s common stock equal to One Hundred and Seventy-Five Percent
(175%) times Note Value divided by the per share price such shares were issued to purchasers of the Company’s equity securities in the IPO rounded to the nearest whole share.
|
|
|
• |
CIC: The Convertible Notes would have automatically converted prior to the effectiveness of such CIC into that number of fully paid and non-assessable shares of the Company’s common stock equal to
Two Hundred Percent (200%) of the Note Value divided by the per share price of the Company’s common stock at which the Company’s common stock was valued in such CIC (after giving effect to such conversion). The Convertible Note holder would
have been entitled to the same contractual rights and would have been bound by the same restrictions and obligations as the other stockholders of the Company in such CIC.
|
|
|
• |
Qualified Financing: The Convertible Notes would have automatically converted into that number of fully paid and non-assessable shares of the Company that were issued by the Company in the
Qualified Financing, determined by dividing an amount equal to One Hundred and Seventy-Five Percent (175%) times the Note Value by the per share price such shares of the Company were issued to purchasers of the Company’s equity securities
in the Qualified Financing, rounded to the nearest whole share. The Convertible Note holder would have been entitled to the same contractual rights and would have been bound by the same restrictions and obligations as the other purchasers
of shares in the Qualified Financing. A Qualified Financing was defined as a sale and issuance of capital stock of the Company (or its successor) in a single transaction or series of related transactions resulting in gross proceeds to the
Company of not less than $5,000,000 (including new equity investment of at least $1,000,000 plus the sum of the outstanding principal amount of the Convertible Notes being so converted under this provision).
|
|
|
• |
Reverse Merger (excluding close of Merger with Rexahn): The Convertible Notes would have automatically converted into that number of fully paid and non-assessable shares of the Combined Company
whose shares were publicly traded in the United States or other jurisdiction following the completion of the Reverse Merger (the “Reverse Merger Parent”), determined by dividing an amount equal to One Hundred and Seventy-Five Percent (175%)
times the Note Value divided by the per share price at which such shares were issued by the Reverse Merger Parent in such Reverse Merger, rounded to the nearest whole share. The Convertible Note holder would have been entitled to the same
contractual rights and would have been bound by the same restrictions and obligations as the other stockholders of the Company in the Reverse Merger.
|
| 6. |
Related Party Transactions
|
| 7. |
Share-based Compensation
|
|
December 31,
|
||||||||
|
2020
|
2019
|
|||||||
|
General and administrative
|
$
|
675
|
$
|
186
|
||||
|
Research and development
|
831
|
122
|
||||||
|
Total share-based compensation
|
$
|
1,506
|
$
|
308
|
||||
|
|
Number of
Options
|
Weighted
Average
Exercise
Price
|
Weighted Average
Remaining
Contractual
Term (years)
|
Aggregate
Intrinsic
Value(1)
(in thousands)
|
||||||||||||
|
Outstanding at December 31, 2018
|
458,219
|
$
|
0.90
|
9.28
|
$
|
26
|
||||||||||
|
Granted
|
579,486
|
$
|
1.19
|
—
|
—
|
|||||||||||
|
Exercised
|
—
|
$
|
—
|
—
|
—
|
|||||||||||
|
Forfeited/Cancelled
|
—
|
$
|
—
|
—
|
—
|
|||||||||||
|
Outstanding at December 31, 2019
|
1,037,705
|
$
|
1.06
|
9.20
|
$
|
1,374
|
||||||||||
|
Granted
|
830,167
|
$
|
3.50
|
—
|
—
|
|||||||||||
|
Exercised
|
(40,672
|
)
|
$
|
—
|
—
|
—
|
||||||||||
|
Forfeited/Cancelled
|
(43,002
|
)
|
$
|
—
|
—
|
—
|
||||||||||
|
Outstanding at December 31, 2020
|
1,784,198
|
$
|
2.17
|
8.87
|
$
|
7,744
|
||||||||||
|
Vested and expected to vest at December 31, 2020
|
895,066
|
$
|
1.23
|
8.14
|
$
|
4,712
|
||||||||||
| (1) |
The aggregate intrinsic value is calculated as the difference between the exercise price of the underlying options and the fair value of our common stock as of December 31, 2020 and 2019 of $6.49 and $2.39 per share (as adjusted for the
Exchange Ratio), respectively.
|
|
2020
|
2019
|
|||||||
|
Expected stock price volatility
|
86.8
|
%
|
92.1
|
%
|
||||
|
Expected life of options (years)
|
7.2
|
5.5
|
||||||
|
Expected dividend yield
|
0
|
%
|
0
|
%
|
||||
|
Risk free interest rate
|
0.6
|
%
|
1.7
|
%
|
||||
|
Number of
Shares
|
||||
|
Non-vested at December 31, 2018
|
64,552
|
|||
|
Granted
|
—
|
|||
|
Vested
|
(64,552
|
)
|
||
|
Non-vested at December 31, 2019
|
—
|
|||
|
Granted
|
40,000
|
|||
|
Vested
|
—
|
|||
|
Non-vested at December 31, 2020
|
40,000
|
|||
| 8. |
Apexian Sublicense Agreement
|
| 9. |
Pre-Merger Financing
|
| 10. |
Net loss per share
|
|
2020
|
2019
|
|||||||
|
Series A and B warrants
|
6,331,674
|
— | ||||||
|
Stock options
|
1,784,198
|
1,037,705
|
||||||
|
Restricted stock awards
|
40,000
|
—
|
||||||
|
Former Rexahn warrants
|
66,538
|
— | ||||||
|
Former Rexahn options
|
123
|
— |
||||||
| 11. |
Income Taxes
|
|
2020
|
2019
|
|||||||
|
Income tax (benefit) provision at federal statutory rate
|
(21.0
|
)%
|
(21.0
|
)%
|
||||
|
Valuation allowance
|
13.8
|
24.2
|
||||||
|
State income tax, net of federal benefit
|
(4.7
|
)
|
(4.7
|
)
|
||||
|
Acquired in-process research and development expense
|
8.8
|
—
|
||||||
|
Warrants
|
7.6
|
—
|
||||||
|
Convertible notes
|
(3.3
|
)
|
1.2
|
|||||
|
Stock options
|
(0.1
|
)
|
0.2
|
|||||
|
Research and development
|
(1.1
|
)
|
—
|
|||||
|
Pass through entity and other
|
—
|
0.1
|
||||||
|
Effective tax rate
|
—
|
%
|
—
|
%
|
||||
|
2020
|
2019
|
|||||||
|
Deferred tax assets:
|
||||||||
|
Federal and state operating loss carryforwards
|
$
|
3,351
|
$
|
1,228
|
||||
|
Acquired intangibles
|
547
|
—
|
||||||
|
Accruals
|
—
|
87
|
||||||
|
Convertible notes
|
—
|
454
|
||||||
|
Organizational costs
|
8
|
9
|
||||||
|
Share-based compensation
|
466
|
81
|
||||||
|
Research and development
|
275
|
—
|
||||||
|
Subtotal
|
4,647
|
1,859
|
||||||
|
Valuation allowance
|
(4,647
|
)
|
(1,859
|
)
|
||||
|
Total deferred tax assets, net of valuation allowance
|
—
|
—
|
||||||
|
Deferred tax liabilities:
|
||||||||
|
Total deferred tax liabilities
|
—
|
—
|
||||||
|
Net deferred tax assets
|
$
|
—
|
$
|
—
|
||||
| 12. |
Subsequent Events
|
|
OCUPHIRE PHARMA, INC.
|
||
|
Dated: March 10, 2021
|
By:
|
/s/ Mina Sooch
|
|
Mina Sooch
|
||
|
President, Chief Executive Officer and Director
|
||
|
By
|
/s/ Mina Sooch
|
Date: March 10, 2021
|
|
|
Mina Sooch
|
|||
|
President, Chief Executive Officer and Director
|
|||
|
By
|
/s/ Amy Rabourn
|
Date: March 10, 2021
|
|
|
Amy Rabourn
|
|||
|
Vice President of Finance
|
|||
|
By
|
/s/ Sean Ainsworth
|
Date: March 10, 2021
|
|
|
Sean Ainsworth
|
|||
|
Director
|
|||
|
By
|
/s/ James S. Manuso
|
Date: March 10, 2021
|
|
|
James S. Manuso
|
|||
|
Director
|
|||
|
By
|
/s/ Cam Gallagher
|
Date: March 10, 2021
|
|
|
Cam Gallagher
|
|||
|
Director
|
|||
|
By
|
/s/ Alan R. Meyer
|
Date: March 10, 2021
|
|
|
Alan R. Meyer
|
|||
|
Director
|
|||
|
By
|
/s/ Richard J. Rodgers
|
Date: March 10, 2021
|
|
|
Richard J. Rodgers
|
|||
|
Director
|
|||
|
By
|
/s/ Susan K. Benton
|
Date: March 10, 2021
|
|
|
Susan K. Benton
|
|||
|
Director
|